
Principal Investigator Training - Role of Principal Investigator in Clinical Research

Have you ever wondered who leads the charge in the battles against diseases, steering the ship through the stormy seas of medical research? Meet the Principal Investigators—unsung heroes whose lab coats barely contain their superhero capes! These rock stars of the clinical world don't just follow the science; they lead it with a baton, orchestrating the symphony that brings us tomorrow's cures. Ready to join the band? Let’s tune up and dive into the world of Principal Investigator Training!
What is a Principal Investigator?
A Principal Investigator (PI) is a crucial team member in clinical research, to some extent, the commander in chief during a battle, except that the battle is fought against diseases and doubts. As a key decision maker, a PI supervises the proceedings of clinical trials in its entirety to guarantee that the study is carried out with the right amount of accuracy, with emphasis on safety and within the context of ethical and regulatory guidelines.
Key Responsibilities of a Principal Investigator in Clinical Research
A Principal Investigator (PI) plays a critical role in ensuring the successful execution of clinical trials. The PI is responsible for maintaining scientific integrity, ensuring regulatory compliance, protecting participant welfare, and overseeing all aspects of data collection and analysis. Below is a detailed explanation of their core responsibilities:
Ethical Oversight: Protecting Participant Welfare
As a Principal Investigator, one of the most important duties is to ensure the ethical conduct of a clinical trial. Since clinical research implies human participants, these guidelines are crucial to follow in order to protect their rights, health, and dignity.
Ensuring Voluntary Participation and Informed Consent
Before enrolling in a trial, every participant must receive comprehensive information about the study, including:
The purpose of the trial
Potential risks and benefits
Alternative treatment options
Their rights to withdraw at any time
This is documented through the Informed Consent Form (ICF), which must comply with ICH-GCP (International Council for Harmonisation - Good Clinical Practice) standards.
Protecting Participant Safety
The PI is responsible for continuous monitoring of patient health throughout the study.
If an adverse event occurs (e.g., an unexpected reaction to a drug), the PI must immediately assess, document, and report it to regulatory authorities and ethics committees.
Ensuring the study adheres to Institutional Review Board (IRB) / Ethics Committee approvals, which oversee participant safety.
Avoiding Ethical Violations
Ethical misconduct, such as coercion, falsified data, or failure to report adverse events, can lead to severe legal consequences.
The PI must enforce transparency and honesty in all research activities.
Regulatory Adherence: Complying with Legal and Institutional Requirements
Clinical trials are strictly regulated to ensure patient safety and data reliability. The PI has to make sure that all the aspects of the trial are compliant with the national and international laws.
Following FDA and ICH-GCP Guidelines
Every clinical trial must follow the ICH-GCP standards, ensuring ethical and scientific quality.
In the U.S., PIs must comply with FDA regulations, including filing an Investigational New Drug (IND) application when testing new drugs.
Other countries follow regulations from authorities like:
EMA (European Medicines Agency)
MHRA (UK Medicines and Healthcare Products Regulatory Agency)
TGA (Therapeutic Goods Administration) in Australia
Submitting Regulatory Documentation
The PI must submit essential trial documents, including:
FDA Form 1572: Declares the investigator’s commitment to following regulations.
Case Report Forms (CRFs): Collects patient data during the trial.
Adverse Event (AE) and Serious Adverse Event (SAE) Reports: Reports any negative reactions from participants.
Adhering to Institutional Policies
If the study is conducted within a hospital or university setting, the PI must ensure compliance with institutional research policies and funding agency requirements.
Data Integrity: Ensuring Accuracy in Data Collection and Analysis
Accurate, reliable and unbiased data collection and analysis is key to the success of a clinical trial, and the PI has overall responsibility for all aspects of data management.
Ensuring High-Quality Data Collection
Every piece of data recorded in the study must be:
Accurate: No discrepancies or data manipulation.
Complete: No missing information.
Verifiable: Supported by medical records and laboratory results.
Preventing Data Fraud
Data fabrication or manipulation is a serious violation that can lead to the invalidation of the entire trial.
PIs must regularly audit the data to detect inconsistencies or irregularities.
Overseeing Data Analysis
Data analysis must be conducted without bias to ensure results are scientifically valid.
The PI must collaborate with biostatisticians to ensure proper statistical methods are used.
Final trial results should be published transparently, contributing to medical knowledge.
Team Leadership: Managing a Multidisciplinary Research Team
A clinical trial is also a team effort - physicians, nurses, data managers, statisticians, and research coordinators are involved. The PI plays the leading role, insuring that the whole team works effectively.
Hiring and Training Staff
The PI is responsible for selecting qualified team members who understand their roles.
Conducting regular training sessions to ensure compliance with:
Good Clinical Practice (GCP)
Standard Operating Procedures (SOPs)
Regulatory requirements
Delegating Responsibilities
A PI cannot perform every task alone. Instead, they must delegate responsibilities to:
Sub-Investigators (Co-PIs): Assist with overseeing different aspects of the trial.
Clinical Research Coordinators (CRCs): Handle patient recruitment and regulatory documentation.
Data Managers: Maintain and verify data integrity.
Facilitating Communication and Conflict Resolution
The PI ensures clear and open communication among all team members.
Addressing conflicts or misunderstandings promptly to maintain a productive research environment.

Why Consider PI Training and Certification?
This is because training and certification for a Principal Investigator (PI) is crucial and necessary for several important reasons. It is not just a question of obtaining a prestigious title: it is a question of thoroughly training oneself to oversee and direct clinical trials that are not only highly complicated and heavily regulated, but also essential for medical science. In the following, I will explain why and how PI training and certification can help professionals who want to do well in this field.
Deepening Knowledge and Expertise
Clinical trials are complex, including multiple processes such as study design, regulatory compliance, participant safety, data management, and ethical considerations. PI training programs provide comprehensive education on these topics, ensuring that PIs are well-versed in:
Clinical Trial Design: Understanding different types of study designs, how to control variables, and how to ensure the reliability and validity of study results.
Regulatory Requirements: Knowledge of global and national regulations such as FDA guidelines, ICH-GCP standards, and others that govern clinical research.
Ethical Conduct: Training in ethical practices ensures that PIs can uphold the dignity, rights, and welfare of study participants.
Such in-depth knowledge is critical not only for the success of clinical trials but also for the advancement of medical treatments and innovations.
Enhancing Credibility and Professional Standing
Certification as a Principal Investigator signals a proven level of expertise and commitment to high standards of clinical research. This credential is recognized across the medical and scientific communities, and can:
Build Trust: Sponsors, regulatory bodies, and participants are more likely to trust a certified PI to conduct studies ethically and competently.
Enhance Resume: Certification can be a significant differentiator in a competitive field, highlighting the PI’s dedication to continuous learning and professional development.
Career Opportunities and Advancement
Trained and certified PIs are in high demand not only in clinical research but also in related sectors such as pharmaceuticals, biotechnology, and academic institutions. The training opens up various career pathways:
Academic Research: Lead cutting-edge research projects at universities or research institutes.
Pharmaceutical Industry: Oversee clinical trials to develop new drugs, working with a team of scientists and regulatory professionals.
Consultancy: Provide expert advice on clinical trial design and management, regulatory compliance, and data integrity.
In these roles, a PI with a strong educational background and a recognized certification can influence the direction of research projects and drive innovations in healthcare.
Meeting the Increasing Complexity of Clinical Trials
As medical science evolves, clinical trials become more sophisticated, testing novel therapies such as gene editing and personalized medicine. Training and certification prepare PIs to handle these complexities by:
Staying Updated: Continuous education through certification programs helps PIs keep up with advancements in technology, treatment modalities, and regulatory changes.
Skill Enhancement: Advanced training modules can include topics like biostatistics, the use of digital tools in data collection, and management of virtual or decentralized trials.
What Does PI Training Cover?
PI training programs typically cover a broad range of topics, including:
Clinical trial design and methodology
Regulatory requirements for clinical research (e.g., FDA regulations, ICH-GCP guidelines)
Ethical considerations in clinical research informed consent, participant protection)
Data management and statistical analysis
Budget development and financial management
Effective communication and collaboration with research sponsors, CROs, and research teams
Benefits of CCRPS PI Training:
Choosing the right training program, like that offered by the Clinical Research Champions (CCRPS), can significantly impact the effectiveness and success of individuals aiming to become Principal Investigators (PIs). CCRPS provides a training program that is not only accredited but also thoughtfully designed to address the complexities of clinical trials management. Below, I will elaborate on the key benefits of enrolling in CCRPS's PI training program, highlighting why it stands out as an excellent choice for aspiring PIs.
Accreditation and Recognition
One of the standout features of the CCRPS training program is its accreditation:
Accredited Curriculum: CCRPS's program is accredited by recognized bodies within the clinical research field. This ensures that the curriculum meets high educational standards, is up-to-date, and is recognized globally.
Professional Validation: Accreditation provides a stamp of quality and reliability, which reassures prospective students that the training they will receive is professionally acknowledged and meets industry standards.
Accreditation enhances the resume of participants and increases their professional credibility in the competitive field of clinical research.
Comprehensive and Updated Curriculum
CCRPS's training program covers all essential aspects necessary for a PI to effectively manage and lead clinical trials:
Broad Coverage: The curriculum includes in-depth training on clinical trial design, regulatory compliance, ethical considerations, patient safety, data management, and statistical analysis.
Current and Relevant: The course material is regularly updated to reflect the latest advancements in medical research and changes in regulatory guidelines. This ensures that participants are learning the most current practices and can apply these effectively in real-world scenarios.
This comprehensive education equips participants with the knowledge and skills to handle the intricacies of clinical trials from start to finish.
Flexibility and Accessibility
The online format of CCRPS’s PI training program offers significant advantages in terms of accessibility and convenience:
Online Learning: The program is delivered online, which allows participants to access high-quality training regardless of their geographical location. This is particularly advantageous for international students or those in remote areas.
Flexible Scheduling: Online courses provide the flexibility to study at one's own pace and according to individual schedules. This is ideal for professionals who may already be working in the field and need to balance training with other responsibilities.
This flexibility makes it possible for a broader range of individuals to pursue PI certification without compromising their current professional or personal commitments.
Preparation for Real-World Challenges
CCRPS’s training goes beyond theoretical knowledge, preparing participants for the practical challenges they will face as PIs:
Practical Applications: The training includes case studies, real-world scenarios, and practical exercises that simulate the day-to-day challenges of managing clinical trials. This hands-on approach helps solidify learning and prepares participants for what to expect in the field.
Skill Development: By engaging with practical components of the course, participants develop critical skills such as problem-solving, decision-making, and effective communication, all of which are essential for a successful PI.
Career Advancement Opportunities
The certification obtained upon completing the CCRPS training program can open doors to numerous career opportunities in clinical research:
Enhanced Job Prospects: Certification demonstrates to potential employers that the individual has undergone rigorous training and possesses the skills and knowledge necessary to manage complex clinical trials.
Professional Growth: The training and certification can lead to career advancement opportunities, including leadership roles within research institutions, pharmaceutical companies, and academia.
Principal Investigator Salary: Compensation and Career Prospects
The salary of a Principal Investigator (PI) in clinical research is influenced by various factors, including experience, location, the type of clinical trials they oversee, and whether they work independently or within an institution. While the role demands a high level of expertise, attention to detail, and responsibility, it also comes with significant financial rewards, particularly for those involved in multiple trials or large-scale research projects.
Salary Range for Principal Investigators
The salary of a Principal Investigator varies significantly based on several factors:
Entry-Level PIs: Physicians or researchers new to the role, with minimal experience, can expect to earn between $80,000 and $120,000 per year.
Mid-Level PIs: With several years of experience and a track record of successfully managing clinical trials, earnings can rise to $150,000 - $250,000 per year.
Highly Experienced PIs: Senior-level PIs with extensive experience, particularly those working in high-demand therapeutic areas (e.g., oncology, cardiology, neurology), can earn $300,000 or more annually, especially if they conduct multiple concurrent trials.
The salary structure can also be influenced by whether the PI works in academia, industry, or as an independent contractor.
Factors Influencing a PI’s Salary
Several elements determine a Principal Investigator’s compensation:
Experience and Qualifications
Medical Degree (MD/DO) vs. PhD: A PI with an MD or DO degree typically earns more than a PhD due to their ability to oversee patient-centered trials and prescribe investigational drugs.
Years in Clinical Research: Senior PIs with 10+ years of experience command higher salaries due to their established reputation and expertise in managing complex trials.
Certifications and Specializations: Holding certifications like the Advanced Principal Investigator Physician Certification (APIPC)™ or ICH-GCP certification can increase credibility and earning potential.
Type and Complexity of Clinical Trials
Phase I vs. Phase III Trials: Clinical trials in Phase I (early drug development) often pay higher due to the increased risks and regulatory oversight compared to Phase III (large-scale efficacy studies).
High-Demand Therapeutic Areas: PIs working in fields such as oncology, cardiology, infectious diseases, and rare genetic disorders can command higher salaries due to the complexity and funding available for these trials.
Multi-Site vs. Single-Site Trials: Managing multiple research sites or working on large-scale multinational studies can lead to higher compensation.
Work Setting
Academic Institutions: PIs working in universities and medical research centers may have a lower base salary ($80,000–$150,000) but often receive research grants, tenure benefits, and funding for publications.
Pharmaceutical and Biotech Companies: Industry-based PIs typically earn higher salaries, often exceeding $200,000, as they manage clinical trials for new drug development.
Independent Research Sites: Some PIs work at dedicated clinical research organizations (CROs), where their compensation is tied to the number of trials they conduct.
Location and Regulatory Environment
United States: PIs in cities with a high concentration of biotech and pharmaceutical companies (e.g., Boston, San Francisco, New York) earn higher salaries due to demand and cost of living.
Europe: Salaries vary widely by country, with Germany, the UK, and Switzerland offering some of the highest pay for clinical research professionals.
Asia and Emerging Markets: Compensation in countries like India and China is lower but increasing due to the growing number of global clinical trials.
Additional Income Sources for Principal Investigators
Beyond base salary, PIs often generate additional revenue streams:
Running Multiple Trials Simultaneously
PIs who oversee multiple clinical trials at the same time can significantly increase their income. For instance, a single study might pay $20,000 to $50,000 per patient enrolled, depending on the trial budget.
Running 3-5 concurrent trials at a time could add between $100,000 to $500,000 per year in additional earnings.
Consulting for Pharmaceutical and Biotech Companies
Experienced PIs often consult for pharmaceutical companies, advising on trial design, regulatory compliance, and patient recruitment strategies.
Consulting fees range from $250 to $500 per hour, depending on expertise and demand.
Grants and Research Funding
PIs in academia may secure grants from agencies like the National Institutes of Health (NIH) or private foundations.
These grants provide additional funding for research, salary support, and professional development.
Publishing and Speaking Engagements
Publishing clinical trial findings in medical journals increases a PI’s credibility and can lead to speaking invitations at conferences, which often come with honorariums of $5,000 to $20,000 per event.
Salary Growth and Career Advancement
PIs can enhance their earnings by:
Becoming Lead PIs on Global Trials: Managing international studies with large patient populations.
Building Research Teams and Centers: Opening clinical research sites to conduct trials independently.
Gaining Specialization in High-Paying Fields: Oncology and rare disease research are among the highest-paying specializations.
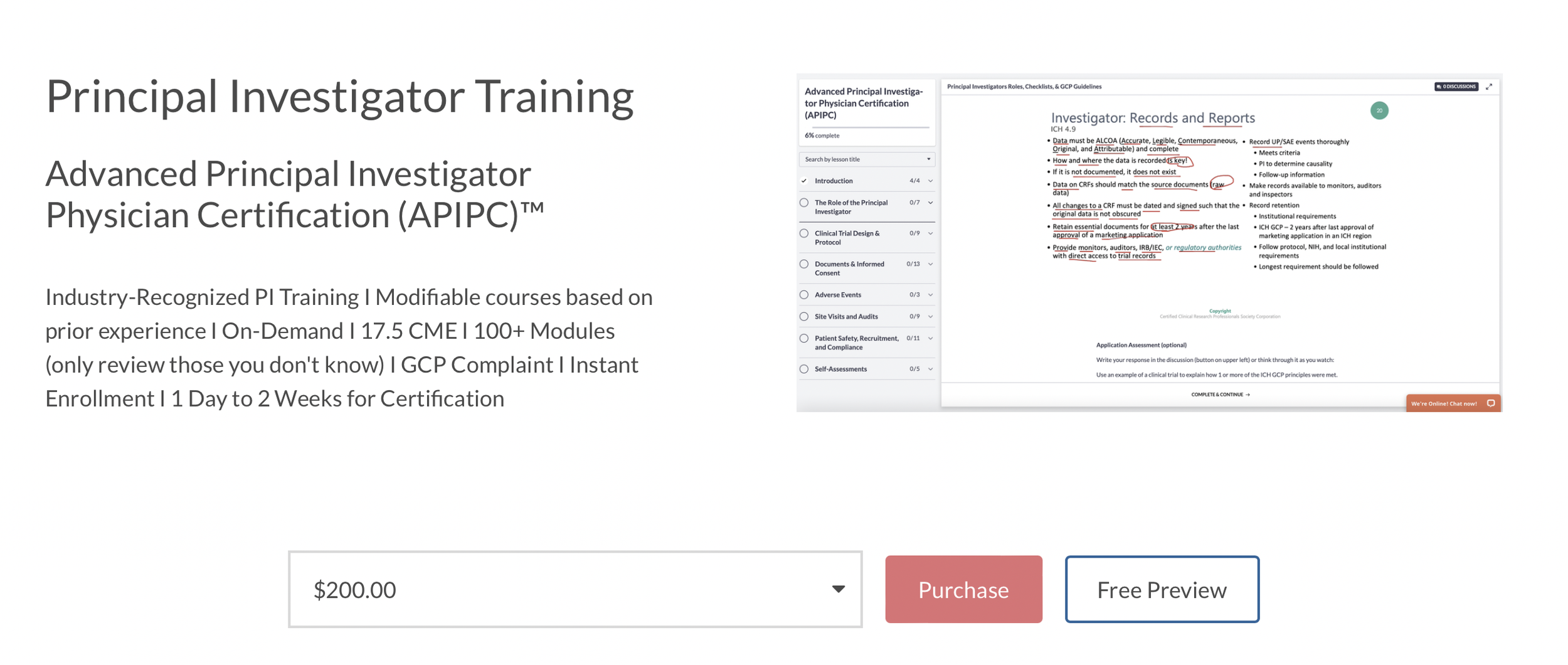
Principal Investigator Training
The responsibility of conducting successful clinical trial lies upon the shoulders of a Principal Investigator. This successful completion of the trials is accomplished by major duties of an Investigator who is responsible for the delegation of duties to other staff members as well as the safety, protection of rights and well-being of the volunteers.
The research volunteers have the right to know about the study that either it is conducted for research purpose or not. Moreover, any alterations in study protocol must be appropriately mentioned to the volunteers and giving them the right to either continue or quit the studies. Providing accurate and appropriate data, informing the ethical committees about the issues related to research site, issues related to adverse effects of drug are the key responsibilities of the Investigator.
To perform all these crucial responsibilities an investigator must pass through the training and evaluation to improve the standards of trials.
The training of PI’s is categorized into different modules so that with the passage of each module all the responsibilities and documents become clear.
PI Course Syllabus
Introduction
Accreditation Statement
CME Handout - How to Obtain 17.5 CME Credits through AMA/ACCME
Principal Investigator Toolkit
How to Effectively Use this Course
The Role Of The Principal Investigator
Principal Investigators Roles, Checklists, & GCP Guidelines
Principal Investigators Reporting Responsibilities for AEs and SAEs
FDA Form 1572 - Part 1
FDA Form 1572 - Part 2
Investigator Initiated Multi-Center Trials
Investigational Product Storage and Dispensing
Investigational Product Accountability in Clinical Trials
Clinical Trial Design & Protocol
Phases of Clinical Trials
Designs of Clinical Trials
Randomized Controlled Trials
Institutional Review Board (IRB)
The Clinical Trial Protocol - Advanced Mastery Review
Protocol Deviations and Violations
Inclusion and Exclusion Criteria in Clinical Research
IND Application
IND and NDA Process
Documents & Informed Consent
Source Documents and Informed Consent Forms
Informed Consent (ICH GCP Section 4.8)
Trial Management, Data Handling, and Record Keeping
Compliance with E-Signatures CFR 21 Part 11
Essential Regulatory Documents Guidance and Binder Tabs (Part 1)
Essential Regulatory Documents Guidance and Binder Tabs (Part 2)
Guidelines for Designing and Completing Case Report Forms
Do’s and Don’ts of a Case Report Form Design
Investigators Brochures
Trial Master File and DIA Model
Trial Master File Reference Guide
Financial Disclosure- Duties and Strategies for Clinical Studies
Financial Disclosures and Conflicts of Interest in Clinical Research
Adverse Events
Advanced Review of Adverse Events
Reporting of Adverse Events
Safety Reporting Requirements for Sponsor Investigators of An IND
Site Visits And Audits
Overview of Types of Monitoring Visits
Site and Investigator Selection
Site Selection/Qualification Visit (Pre-Study Visit)
Site Close Out Visit
Audits vs. Inspections
FDA Warning Letter
Site FDA Audit Inspection Checklist
How to Survive Through an FDA Inspection
Do and Don’ts during an FDA Inspection
Patient Safety, Recruitment, And Compliance
Introduction & History of ICH GCP
Compliance Requirements in Clinical Trials
Subject Recruitment and Retention (Part 1)
Subject Recruitment and Retention (Part 2)
Safety of Human Subjects in Clinical Research
Ethics of Research Involving Pregnant Women and Fetuses
Ethics of Research Involving Mentally Incapacitated
Ethics of Research Involving Children
Scientific Misconduct in Research and How to Prevent It
Increasing Subject Compliance in Clinical Trials
Misconduct in Research – Detecting Falsification
Self-Assessments
Self-Assessment MiniQuiz 1
Self-Assessment MiniQuiz 2
Self-Assessment Quiz A
Self-Assessment Quiz B
Final Quiz
The Drug Development Process for PIs:
In the first module of training, PI is introduced with the clinical research trials, phases, pre- clinical research, drug developing method and dealing with common difficulties. PI must be aware of the purpose of clinical trials which are conducted to develop a drug to ultimately leading to the capable the drug used for prevention, diagnosis and treatment of disease.
The main objectives of the clinical trials include:
Treatment is the common fascination for the subjects to participate in the trials as
it provides them with medicine for treatment and diagnosis.
Observational studies enable the investigator to find the relationship between the
habits of socioeconomic group and the progress pattern of disease.
Prevention type of studies include subjects who want to prevent the disease rather
than treatment. Such kind of studies require the personnel having family history of
the disease.
Diagnostic type of trials is used to treat the disease efficiently.
Basic science by its nature allows to concentrate upon the phenomenon, closed
observation, experimentation of hypothesis, extracting conclusion to develop a new approach.
Pre-clinical trials as the name indicates are performed before the clinical trials and commonly known as Phase 0 trials involve First-in-man trials to know about the safety of the drugs and allows the determination of amount of safe dose of drug. This kind of trial is used to test the new medical devices, diagnostic tools, gene therapy and drugs.
There are mainly 4 phases of clinical research trials.
Phase 01. Determination of pharmacology and tolerability
Phase 02. Evaluation of safety and efficacy
Phase 03. Evaluation of safety and risk ratio as well as effectiveness. Successful completion of phase 3 trials leads to submission of application for the FDA approval.
Phase 04. Monitoring long term effects and effectiveness. Successful phase 4 trials are considered as FDA approved drug.
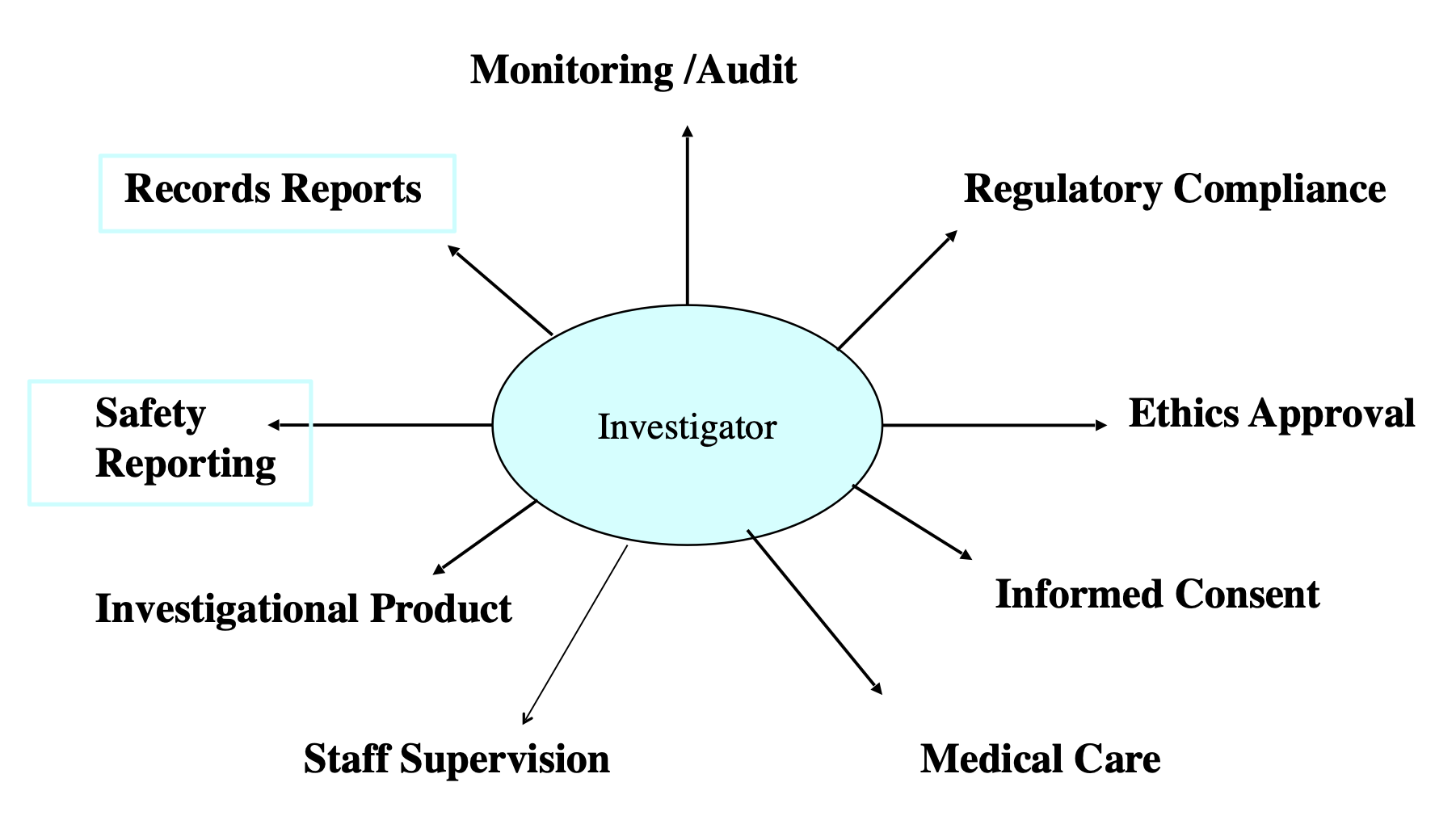
The Role of Principal Investigator:
A person responsible for the conduct, preparation, research grants, agreements, training or public service project, contract or other sponsored projects in compliance with applicable laws and regulations and policy governing the conduct of sponsored research.
FDA recommends a form duly filled and signed by the investigator which serves as a statement from the side of investigator is known as FDA form 1572
Major Roles of PI:
The major roles of the PI are The Position roles (managing the integrity of design and collaborative results, reporting to the individuals like dean , department head and divisional chief),coordination with the administration, scientific proposal preparation, arranging and managing budgets, preparation of protocol according to the regulations and assuring the approval of any changes before implementation, acceptance of award in the light of rules and regulations, Conducting research (supervision to implement the rules, supervision of the team to ensure ethical conduct, reporting any misconduct), applying appropriate cost rate to the facilities and administrative, allocation of space to conduct the research, arranging equipment for study etc.
ICH has provided adequate GCP for Investigator to work effectively. These guidelines provide information about following aspects:
Qualification and dealing with the agreements.
Adequate resources in terms of staff and budget.
Protection of the rights and safety of trials subjects either human or animals.
Procedure of communication with regulatory bodies like IRB/IEC.
Following and implementation of the rules and protocols set by regulatory bodies,
Allowing the supervision of the investigational product to a qualified and responsible
person.
Responsibilities regarding design of studies (randomization and blinding).
Arranging the informed consent of individuals.
Keeping records and reporting to the regulatory bodies.
Safety reporting
Termination or suspension of the trials before time.
Submission of final reports.
Informed Consent:
A document signed by every volunteer participating in the trial by informing him /her regarding all the matters of trials like, either he /she is voluntarily participating in the trials, informed about the risks, clarifying the role in trials and procedure of study. Federally, it is compulsory to get signed informed consent before participation in the clinical trials.
This link provides all the information regarding inform consent form and ICF review sample.
To effectively complete the consent process, interviewer should provide sufficient time to the participant to think that either he should participate or not. This process also allows the participants to review the procedure and frequently ask any query related to the trials. This process must be transparent and free of influences.
For developing a consent form standard UCI, IRB consent template must be followed. It must include the following key points.
General information regarding purpose, design, and statement of study.
Objective of the study must be clear including an appropriate definition that why
FDA testing is necessary.
Complete description of the process either in tabular or descriptive form comprising
of complete description of stages involved in process, inclusion/ exclusion criteria,
type of questions in case of questionnaire.
Risks associated with the study must be properly defined in tabular or descriptive
form. If risks are not clear, then a statement must be added that any unforeseeable
risk can occur.
To clarify the participant can withdraw or Investigator can terminate any participant
at any time. It is investigator’s responsibility to clarify the procedure of the
withdrawal from the study when participant wants.
Confidentiality must be maintained regarding the subject’s information, procedure,
and research data and about the record keeper.
Investigator will provide new information to the subject by standard text.
Or the benefit of the participant who wants to withdraw from the studies, extra
courses or benefits must be explained in an appropriate way.
Research member’s financial interest in the study must be mentioned as well as the
specimen collection should be informed.
It must be clearly written that the subject has the right to either participate or not in
the study.
Signature line for participant and authorizing member must be included. Witness
line shall also be included in some kind of studies where necessary.
The Clinical Research Protocol:
Protocol is the document describing the procedure of conducting clinical trials ensuring the safety of subjects or volunteers and integrity of collected data. This document must comprise of following headings and details regarding trials:
Title page
Background information
Objective /purpose
Study design
Inclusion and exclusion of subjects
Treatment of subjects
Evaluation and assessment of safety and efficacy
Adverse events
Suspension of study
Statistical data
QC and QA
Ethics
Data handling and record keeping
Publication policy
Project timetable
References
Appendices
The NIH also provides templates and resources to develop the protocols and designs to conduct the trials.
When the activities without any significant reason diverge from the IRB approved protocol the is known as protocol deviation.
Adherence to the trial related requirements, following GCP requirements, and fulfilling regulatory requirements is known as Protocol compliance.
Understanding Adverse events:
Adverse event is defined as an unintended and unwanted sign, symptom or disease partially associated with the use of drug without any judgement about causality and relationship to the drug.
If a sponsor or investigator based on outcomes (death, life threatening adverse reaction, In-patient hospitalization or disruption life functions) considers the adverse event or reaction serious.
All adverse events should be documented in the patient’s medical record. To collect AE’s patients should not be asked anticipated questions either they should be asked the open- ended questions, during examination and evaluation. A progress note must contain good clinical practice and good clinical practice research. Report must contain date of AE initiation, attribution of AE, date of resolved AE, documentation of worsen or untreated AE.
Recording of AE onto a case report form (CRF) includes details:
Date of initiation of AE
Treatment of AE
Garde of severity
Attribute of AE
AE resolved date.
For worsen condition or treatment changing progress or any relationship should be
documented on the other CRF form
Reporting to regulatory bodies involves routine and expedited.
IND Safety reports (ISR) comprised of all types of adverse events, In vitro testing details, findings of other studies and all other suspected adverse events.
Format for submission of an ISR include Narrative ( all kind of data either published or unpublished analysis data),
FDA form
Council of International Organization Medical Sciences Form
Source Documentation
A pharmaceutical company or sponsor provide or sends the new drug for approval to the FDA including all the data of clinical trials. The FDA then asks for the provision of the source data from where it was captured. This documented data is called source document.
Essential documents in clinical trials
Investigator’s Brochure, study protocol, Informed consent or subject information, reports of research trials and case report form.
Firstly, Investigator brochure (IB) contains information about the investigational drug before and after the performing he clinical studies in a brief and concise manner. This document is comprised of keywords and abbreviations, contents list, brief description of the investigational drug, general approach towards study and brief description as introduction, characteristics of the medicinal product, nonclinical and clinical studies and at the end conclusion and references.
Secondly, Clinical Study Protocol is a documentation of goals, objective, and design of clinical study. This document is designed after the instructions of all parties participating in clinical trials and this document should contain all the information regarding clinical trials and then sent to the authorities for review.
Thirdly, when any amendment is made in the study protocol, protocol amendment document is used. Before implementation it must be again approved by the authorities.
Fourth, Informed consent is the document to ensure that the volunteers are perfectly aware of the objective of trials, Investigational product. This document depends upon the willingness of the volunteers and they have right to leave the trials.
Fifth, Study progress reports are prepared by the medical monitor either on daily basis or final report is prepared to show the committees.
Sixth, Case record form is the document to record the data of individuals involved in the study. It could either be an electronic document.
Responsibilities of an investigator according to GCP guidelines include:
Supervision of the conduct of clinical investigation.
Delegation of the duties to the qualified personnel.
Training of the participating staff
Arranging an individual for the supervision of each site.
Protection of rights, safety and welfare of study objects.
Communication with IRB requires high level of professionalism for which PI assigns this duty to Regulatory document specialist (RSD) and documentation of this communication is maintained.
SOP’s for communication are provided in this link.
Deviation’s fraud and noncompliance:
When IRB or any committee sets regulations, rules, policies, and laws and rely upon the organization or any person to imply them, then any failure in implementation of these rules is commonly described as noncompliance. To overcome this issue, NLM has created a website and provided the time frame to government and private trials conducting bodies to register and provide the results within the given time frame.
A term fraud is commonly used in clinical trials which means contravention of faith and dealing intentionally, to harm any individual by manipulating the research data and results. It also includes deviating from the set protocols, policies and manipulating data and research results. The question arises that how and why fraud occurs in clinical trials. There are some points summarized to explain it.
To gain fame by participating in internationally renowned trials.
Manipulating data of repeated testing due to lethargy of research staff or
investigator.
Rule’s policies and incentives attached to the trials are the environmental factors.
Idiosyncrasies, ego, competition among colleague investigators, for increasing the
tenure and promotion.
When an investigator either intentionally or unintentionally fails to comply with the policies and regulatory requirements, FDA has authority to disqualify him either permanently or temporarily. This disqualified investigator is not allowed to conduct any kind of investigations regulated by FDA.
The main steps or headings of disqualification of an investigator by FDA are described here.
• The Disqualification processes.
o Issuance of notification for disqualifying an investigator and providing an opportunity to explain.
o Consent agreement
o Summon of hearing as an opportunity after disqualification. o After hearing taking crucial decisions
o Actions upon disqualification:
Criminal prosecutions
Revealing the information regarding decision of disqualification
Reinstatement of disqualified investigator
The link for the detail study: FDA proceeding
10 Lesser-Known Facts About Principal Investigators in Clinical Research
Non-MDs Can Be Principal Investigators – While most PIs are physicians, PhDs, PharmDs, and other researchers can also lead trials, provided a licensed physician is a Co-Principal Investigator in trials involving direct patient care.
PIs Can Earn Royalties from Research – If a PI's research leads to a patented drug or medical device, they may receive royalties or licensing fees, making clinical research a lucrative field beyond trial funding.
Clinical Trials Can Pay PIs Per Enrolled Patient – In some trials, PIs earn between $3,000 to $10,000 per patient enrolled, meaning higher recruitment = higher earnings.
Some PIs Work as Independent Contractors – Instead of working for hospitals or pharma companies, some freelance PIs work on a contract basis, running trials at multiple research sites, increasing income flexibility.
PIs Are Legally Responsible for Their Trials – If a study violates ethical guidelines or results in patient harm due to negligence, the PI can face lawsuits, disqualification, or regulatory action by agencies like the FDA or EMA.
A Single Protocol Violation Can Invalidate an Entire Trial – Even a minor deviation from trial protocol (such as missing documentation or incorrect patient dosing) can jeopardize FDA approval and require costly remediation.
Most PIs Have to Secure Their Own Trial Funding – While pharmaceutical companies fund many trials, academic and independent PIs often have to apply for NIH, private, or institutional grants to support their research.
Medical Monitors Often Earn More Than PIs – Some experienced PIs transition into roles as Medical Monitors, overseeing multiple clinical trials globally for higher salaries ($200K-$400K annually) while avoiding patient care responsibilities.
Regulatory Inspections Can Happen at Any Time – The FDA or other regulatory bodies can conduct surprise inspections at a PI’s research site to check compliance, and any serious issues can lead to trial shutdowns.
PIs Play a Role in Drug Pricing & Approval Decisions – The data collected by PIs doesn’t just determine whether a drug is effective—it also influences its market price, insurance coverage, and regulatory approval speed.
Conclusion
A Principal Investigator plays a crucial role in clinical research, ensuring ethical standards, regulatory compliance, and successful trial execution. With the growing demand for qualified PIs, the right training and certification can set you apart in academia, pharmaceuticals, and global clinical trials.
At CCRPS, we offer the best Principal Investigator Certification, equipping professionals with the expertise needed to lead groundbreaking research. Take the next step in your career and shape the future of healthcare today.
Frequently Asked Question (FAQs)
What qualifications are required to become a Principal Investigator in clinical research?
To become a Principal Investigator (PI), you typically need:
A medical degree (MD, DO) or a PhD in a relevant scientific field.
Clinical research experience, usually gained through working on trials as a Sub-Investigator (Sub-I) or Clinical Research Associate (CRA).
Knowledge of Good Clinical Practice (GCP), FDA regulations, and ICH guidelines.
Principal Investigator training or certification, such as the Advanced Principal Investigator Physician Certification (APIPC)™ from CCRPS.
Some non-MD professionals can become PIs if they work under a Co-Principal Investigator who is a licensed physician.
How does Principal Investigator training improve clinical trial outcomes?
PI training ensures:
Regulatory Compliance – Understanding FDA, ICH-GCP, and IRB/IEC requirements.
Patient Safety – Proper handling of adverse events and ethical trial management.
Data Integrity – Ensuring accurate, complete, and verifiable research data.
Trial Efficiency – Effective protocol design, team leadership, and resource management to prevent trial delays or errors.
Proper training leads to higher-quality clinical research, reducing risks and improving trial success rates.
Is Principal Investigator certification mandatory for conducting clinical trials?
No, certification is not legally required, but it is highly recommended. Certification:
Enhances credibility and trustworthiness with sponsors, CROs, and research institutions.
Increases job opportunities and funding prospects.
Provides essential regulatory knowledge to ensure compliance with trial protocols.
Helps first-time PIs gain the confidence and skills needed to lead trials successfully.
While experience is a key requirement, certification gives you a competitive advantage.
What are the biggest challenges faced by Principal Investigators?
PIs handle various challenges, including:
Regulatory Compliance – Keeping up with evolving FDA and ICH-GCP guidelines.
Patient Recruitment & Retention – Ensuring enough participants while maintaining diversity.
Budget & Resource Management – Managing trial costs and sponsor expectations.
Data Accuracy & Integrity – Preventing errors, fraud, or protocol deviations.
Time Management – Balancing clinical duties, research, and administrative tasks.
Strong leadership, proper training, and a well-structured team help PIs overcome these challenges effectively.
How long does it take to complete Principal Investigator training?
The duration varies based on the program and the trainee’s background. On average:
Online PI training courses, like those from CCRPS, can be completed in 2-6 weeks at a flexible pace.
In-person training and workshops may take several months.
Full certification, including experience requirements, may take 1-2 years.
Most experienced physicians or researchers can complete PI training quickly, while new investigators may need additional time for practical application.
Can a non-MD become a Principal Investigator in clinical research?
Yes, non-MD professionals such as PhDs, PharmDs, and other researchers can become PIs in certain clinical trials. However, some restrictions apply:
If a study involves drug administration, medical procedures, or patient treatment, a licensed physician (MD/DO) must be a Co-Principal Investigator.
Non-MD PIs often lead observational or laboratory-based trials where patient treatment isn’t required.
Each study's requirements depend on institutional policies, regulatory bodies, and trial design.
What is the difference between a Principal Investigator and a Sub-Investigator?
Principal Investigator (PI):
Has full responsibility for the study’s design, execution, and compliance.
Oversees participant safety, trial integrity, and regulatory adherence.
Leads the research team and communicates with sponsors and regulatory bodies.
Sub-Investigator (Sub-I):
Assists the PI with specific tasks like patient monitoring, data collection, or drug administration.
Reports to the PI and helps ensure trial efficiency and compliance.
Often serves as a stepping stone toward becoming a PI.
While both roles are crucial, the PI holds ultimate responsibility for the trial’s success.
How can Principal Investigators increase their earnings in clinical research?
PIs can maximize their salary and income potential by:
Running Multiple Trials: Conducting 2-5 trials at a time can significantly boost earnings.
Specializing in High-Demand Therapeutic Areas: Oncology, neurology, and rare diseases offer higher compensation.
Consulting for Pharmaceutical Companies: Providing expert guidance on trial design and regulatory compliance.
Becoming a Medical Monitor: Overseeing trials for sponsors and CROs.
Obtaining Certifications & Advanced Training: Earning credentials like the APIPC™ can increase credibility and job opportunities.
PIs who manage large-scale trials or work in high-demand locations often earn six-figure salaries or more.
{kind=link}