
An In-Depth Guide to Form FDA 1572

Form FDA 1572, officially known as the Statement of Investigator, is a legally binding document required by the United States Food and Drug Administration (FDA) for clinical trials conducted under an Investigational New Drug (IND) application. This form ensures that clinical investigators comply with FDA regulations, maintain ethical standards, and uphold the rights, safety, and welfare of trial participants. Many researchers often search for "What is FDA Form 1572 used for?" or "Who is required to sign Form FDA 1572?" to understand its purpose and necessity.
The FDA mandates that any investigator participating in a clinical trial under an IND must sign Form FDA 1572 before the study begins. The investigator must agree to comply with regulatory requirements, follow Good Clinical Practice (GCP) guidelines, and ensure that the rights and safety of human subjects are protected. Since compliance is crucial, understanding how to complete Form FDA 1572 correctly and avoid common mistakes is essential for investigators and sponsors alike.
What is the Form FDA 1572?
The Form FDA 1572 is a form issued by the United States Food and Drug Administration (FDA). It is a document that must be completed and submitted by sponsors and investigators conducting clinical trials in the United States. This form provides the FDA with important information about the clinical trial, such as the protocol, investigator qualifications, and other important information.
CCRPs provides principal investigator certification to ensure accuracy and efficiency for form completion and compliance.
Purpose and Importance of Form FDA 1572
Form FDA 1572 serves as a legal contract between the investigator and the FDA. It establishes the investigator's responsibility to adhere to the approved study protocol, maintain accurate records, and report significant findings. Failure to comply with these regulations can lead to FDA inspections and potential penalties. Many investigators often search "How often should Form FDA 1572 be updated?" because any major changes require resubmission of the form to maintain compliance.
Additionally, this document helps the FDA monitor investigator qualifications, study conduct, and research integrity. The form also provides a crucial compliance check for Institutional Review Boards (IRBs) by ensuring that investigators have the appropriate credentials and facilities to conduct the study safely.
Who Must Complete Form FDA 1572?
Clinical investigators conducting trials under an IND application must complete Form FDA 1572. This includes Principal Investigators (PIs), sub-investigators, and clinical research staff involved in the study’s execution. Many professionals ask, "Can a non-physician be a principal investigator?" The answer is yes, as long as they possess the necessary training, experience, and qualifications to oversee a clinical trial.
Form FDA 1572 is not required for studies not conducted under an IND. However, compliance with GCP guidelines is still recommended to ensure research integrity and patient safety. Investigators involved in trials outside the U.S. frequently search, "Does Form FDA 1572 apply to non-IND trials?" to confirm their responsibilities under different regulatory conditions.
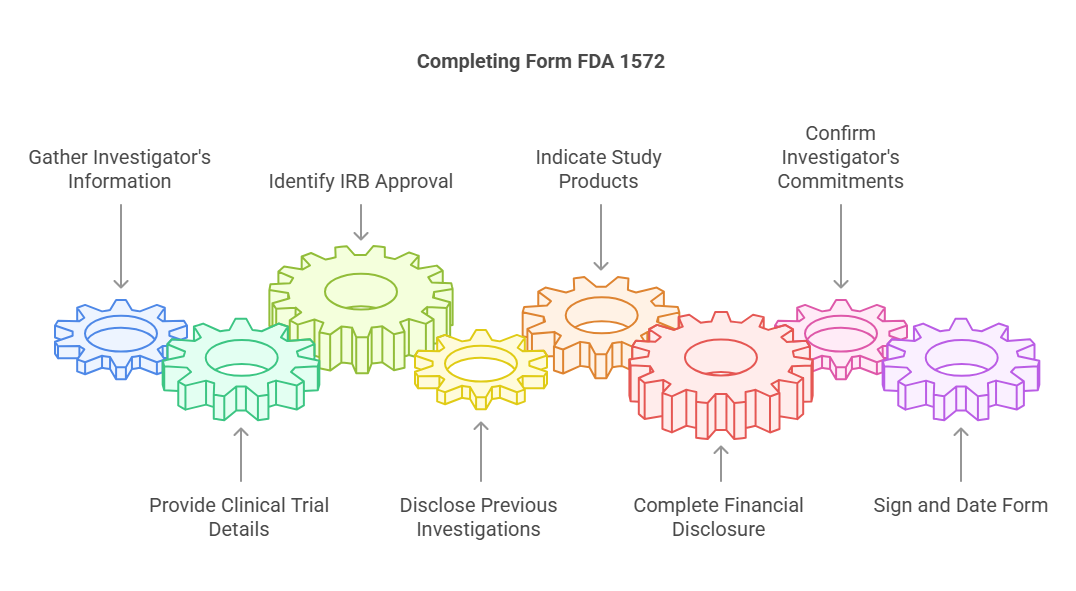
How to Complete Form FDA 1572: Step-by-Step Guide
Filling out Form FDA 1572 correctly is essential for regulatory compliance. Below is a breakdown of each section:
Investigator’s Information: This section requires the investigator’s full name, office address, phone number, and email. Ensuring the accuracy of this information is vital, as errors could delay the approval process.
Clinical Trial Details: Investigators must provide the protocol number and title of the clinical study. Additionally, they should list the name and address of the facility where the research will take place.
Institutional Review Board (IRB) Approval: The IRB overseeing the study must be identified, including its name and contact details. The IRB ensures that the study is ethically conducted and protects the rights of trial participants.
Previous FDA-Regulated Investigations: Investigators should disclose prior research conducted under FDA oversight. This includes listing IND numbers and investigational products used in past studies.
Drugs or Devices Used in the Study: Investigators must indicate whether the study involves drugs, biologics, or medical devices. They should list all investigational products and their intended dosage regimens.
Financial Disclosure: This section requires investigators to disclose any financial interests related to the study. Avoiding conflicts of interest is crucial to maintaining research integrity.
Investigator’s Commitments: By signing Form FDA 1572, the investigator agrees to follow the study protocol, ensure proper data collection, and comply with FDA regulations regarding informed consent and patient safety.
Signatures and Date: The principal investigator must sign and date the form before submission. If any information changes, an updated form must be submitted.
Common Mistakes to Avoid
Many researchers ask, "How to correct errors on an already submitted FDA 1572?" If an error is detected after submission, the investigator should submit an amended form with the correct information. Here are some common mistakes to avoid:
Leaving Sections Incomplete – Missing details can delay approval.
Providing Inaccurate Information – Always verify data before submission.
Failure to Update the Form – Any significant changes require a new submission.
Neglecting to Report Financial Conflicts – Transparency in financial interests is required.
Key Differences Between FDA 1572 and Non-IND Research Documentation

Recommended Clinical Research Courses
To enhance compliance and efficiency in clinical research, consider enrolling in certification programs:
Final Thoughts
Form FDA 1572 is a critical document in FDA-regulated clinical trials. Ensuring that it is accurately completed and submitted is essential for regulatory compliance and participant safety. By following this guide, investigators and sponsors can streamline their processes and avoid compliance pitfalls. For further training, explore CCRPS courses and enhance your expertise in clinical research.
FAQs
What is the Statement of Investigator, Form FDA 1572?
The Statement of Investigator, Form FDA 1572, is a document that must be completed and signed by the lead investigator for each clinical investigation conducted under an Investigational New Drug Application (IND). It is used to provide information about the qualifications of investigators conducting studies with investigational drugs.
Why does this form need to be completed by an investigator?
This form needs to be completed by an investigator to ensure that they are qualified and have the necessary experience and expertise to conduct a safe and ethical clinical trial. This form also serves as affirmation from the investigator that he or she has read and understood the protocol of the clinical investigation in question, as well as any other information pertinent to the study provided by the sponsor or sponsor-investigator.
When must this form be completed and signed by an investigator?
The form must be completed and signed by an investigator at or before initiation of a clinical investigation which involves use of an investigational drug. The form must also be updated or a new 1572 must be completed and signed by an investigator if there is new or changed information relevant to the study.
Must the investigator be a physician? What are the minimum qualifications of an investigator?
An investigator does not need to be a physician, but should meet certain criteria set forth by FDA such as having sufficient training, knowledge, and experience pertinent to the type of research being conducted; having access to medical records relevant to studies being conducted; understanding good clinical practice requirements; following protocols; and obtaining informed consent from research participants.
Does the 1572 need to be submitted to FDA?
Yes, this form needs to be submitted to FDA along with supporting documents prior to initiation of a clinical trial involving use of an investigational drug. Even if a foreign clinical study is not conducted under an IND, investigators who conduct such studies still may need to sign a 1572 in certain circumstances.
If a clinical investigation is not conducted under an IND or is for a medical device, must investigators sign a 1572?
A sponsor may conduct a foreign clinical study under an IND only in situations where it does not qualify for exemption from IND regulations due to lack of assurance that subject protection will be maintained without oversight from FDA. If such conditions are met then sponsors must submit an IND application prior initiating the foreign study in order for it to comply with applicable regulations.
Must investigators who conduct studies outside of the United States sign a 1572?
Yes, according to the Food and Drug Administration (FDA), all clinical investigators conducting studies on FDA-regulated products that require an Investigational New Drug (IND) application must sign a Form FDA 1572. This form is used to confirm that the investigator understands their obligations and responsibilities related to conducting IND-related studies.
If a foreign clinical study is being conducted under an IND, what are the investigator's responsibilities with respect to local laws and regulations?
When conducting foreign clinical trials under an IND, investigators must comply with both local laws/regulations as well as those set forth by the FDA in 21 CFR Part 312. This includes ensuring that good clinical practice standards are followed and that any applicable ethical considerations are taken into account when designing and implementing the study protocol. In order to ensure compliance with local laws, investigators may need to obtain permission from national or regional regulatory authorities before beginning the trial. Additionally, depending on the country in which a foreign clinical trial is conducted, additional requirements such as language translations of informed consent forms may be necessary.
For foreign clinical studies conducted under an IND, how can an investigator sign the 1572 when he/she knows he/she cannot commit to all of the requirements on the form, specifically IRB membership (21 CFR 56.107)?
In order for an investigator to sign a Form FDA 1572 for a foreign clinical study under an IND even if they know they cannot commit to all of its requirements (specifically IRB membership), they should discuss this issue with their sponsor prior to signing it in order to find out what alternative arrangements can be made. Furthermore, sponsors should consider both local laws/regulations as well as ICH standards when making these arrangements so that appropriate safety measures can be taken. For instance, sponsors may choose to contract independent consultants or external experts who are familiar with good clinical practice standards in order to review data gathered during trial activities at sites located outside of United States jurisdiction.
If a sponsor chooses to conduct a foreign clinical study (or operate non-US sites in a multinational study) under an IND and the investigators at these non-US sites comply with ICH E6 Good Clinical Practice Consolidated Guidance, would the non-US investigators also be in compliance with FDA's IND requirements under 21 CFR Part 312?
When conducting foreign clinical trials under an IND, compliance with ICH E6 Good Clinical Practice Consolidated Guidance alone may not guarantee full compliance with 21 CFR Part 312 requirements set by the FDA. Although ICH standards provide general guidance on how research should be conducted ethically and safely within different jurisdictions around world, some countries have rules or regulations in place which differs from those established by ICH E6 Good Clinical Practice Consolidated Guidance or which might amend them slightly; therefore potential discrepancies between these two sets of regulations need to be taken into consideration when designing trial protocols for international trials subject to FDA jurisdiction. Furthermore, sponsors should ensure that all parties involved in such trials understand their individual responsibilities related executing Research Ethics Committee approval processes required for each country included in study protocol design prior commencing trial activities at each site outside US jurisdiction
Must foreign clinical study sites in a multinational study that includes domestic sites be conducted under an IND?
Yes, all foreign clinical study sites that are part of a multinational study must be conducted under an IND. The sponsor must submit an application to the FDA for approval to conduct the study and provide detailed information about the site, such as personnel qualifications, resources and facilities available at the site, and protocol for conducting the research. The IND application includes protocols and other information describing how a proposed clinical investigation will be conducted.
How does a sponsor submit information to FDA about a foreign clinical study that was not conducted under an IND?
The sponsor must submit an Investigational New Drug (IND) Application to the FDA if they wish to conduct a foreign clinical study which has not been previously approved by the FDA. The sponsor should include detailed information regarding the proposed clinical trial, including the proposed protocol, safety measures put in place to protect subjects participating in the trial, qualifications of personnel involved in conducting or supervising the trial, and any other information which will help demonstrate compliance with applicable regulations.
Should a new form be prepared and signed when the OMB expiration date is reached?
No, there is no need for sponsors to prepare or sign any new forms when submitting an Investigational New Drug (IND) Application or when seeking approval from FDA for any particular clinical trial. However, sponsors must follow all applicable laws and regulations related to their research activities and comply with requirements set forth in relevant documents such as Form 1572 (Declaration for Clinical Investigations Involving Human Subjects), Form 3454 (Statement of Investigator), and Form 3753A (Clinical Investigator's Brochure).
Does FDA expect a double-sided 1572, or is a two-page document printed from the FDA website acceptable?
The FDA requires sponsors to submit Form 1572 as part of their IND application as both single-sided copies and double-sided copies. The form should be completed according to applicable regulations outlined by 21 CFR 312.23(a)(7). Sponsors may not use double-sided copies of documents obtained from websites hosted by other organizations, including those belonging to different government agencies or non-profit institutions..
How should the 1572 be completed?
Form 1572 should be filled out completely by each investigator listed on it who is responsible for conducting or supervising certain aspects of research activities at any given site. This includes providing all necessary details such as person’s name, address/location(s), contact information (e-mail address/phone number/fax number etc.), signature(s) etc., along with listing any degrees/licenses held by him/her that show he/she is qualified to conduct/oversee said research activities being funded through this particular project. Furthermore important section detailing ‘Financial Disclosure’ needs special attention especially since this form also serves purpose of informing potential participants about potential conflicts of interest pertaining to investigator’s involvement in these studies alongside his/her salary details etc. So it is crucial that this section is filled out completely without leaving out any significant details so that true picture can be presented in front of future volunteers who might decide whether they want participate in said studies or not based on aforementioned disclosure
Review Questions for FDA Form 1572
What is FDA Form 1572?
A) A form that must be completed and signed by the clinical investigator when a study is initiated, revised, or discontinued
B) A form that must be completed by all patients participating in a study
C) A document used to report adverse drug events to the FDA
D) A document used to collect information about the safety and effectiveness of drugs
Answer: A) A form that must be completed and signed by the clinical investigator when a study is initiated, revised, or discontinued. Explanation: The FDA Form 1572 is an agreement between investigational sites and the FDA. It outlines key elements of studies conducted at those sites such as background qualifications of investigators and staff, source documents, records maintenance, reporting requirements and procedures for handling drugs used in clinical trials.
What type of information must be provided when completing FDA Form 1572?
A) Personal information about each individual participant in a trial
B) Information about drugs being tested in a trial
C) Financial information from sponsors involved in the trial
D) Information about laboratory tests performed during the trial
Answer: B) Information about drugs being tested in a trial. Explanation: The FDA Form 1572 requires that the investigator identify all drugs to be administered during the investigation (e.g., active ingredient names and doses), along with any other products that may affect laboratory results such as vitamins or minerals. This will help ensure accurate record keeping throughout the trial.
Who is responsible for ensuring accuracy on FDA Form 1572?
A) The clinical investigator conducting the study
B) The sponsor of the study/trial
C) The patient participating in the study/trial
D) All of the above
Answer: D). All of the Above. Explanation: Accuracy on FDA Form 1572 is essential since it serves as an agreement between investigational sites and the Food & Drug Administration (FDA). Thus, both sponsors and clinical investigators are responsible for ensuring accuracy on this form, as well as patients who participate in studies/trials should they provide any data or information required by this form.
When does an individual need to submit an updated version of FDA Form 1572?
A) When enrolling new patients into a clinical trial
B) When changes are made to protocols related to a given clinical trial
C ) When making changes to personnel associated with a given clinical trial
D ) All of the above
Answer: D). All of the Above Explanation: An updated version of FDA Form 1572 needs to be submitted when enrolling new patients into a given trial; when changes are made to protocols related; or when personnel associated with a given clinical trail have changed since its initiation or last update. This helps ensure accuracy so that all parties involved have access up-to-date information regarding ongoing studies/trials they’re involved with at any given time.
What happens if an individual fails to submit an updated version of FDA Form 1572?
A ) They will not receive funding for their research project
B ) Their research project may not pass inspection from regulatory authorities
C ) They may face legal repercussions from regulatory authorities
D ) All of the Above
Answer: D). All of The Above Explanation: If an individual fails to submit an updated version of FDA Form 1572 then they can face various consequences such as not receiving necessary funding for their research project; having their research project fail inspection upon review by regulatory authorities; or facing legal repercussions from said authorities due its importance in providing complete documentation related to ongoing studies/trials involving human subjects which helps protect participants’ rights while conducting necessary research work safely and ethically within regulatory guidelines set forth by law enforcement bodies responsible for protecting public health around world according these standards set forth through years long process establishing best practices medical community has come accept today across many countries globally depending respective jurisdiction laws apply under question particular case being consider review possible action taken based findings presented within scope parameters policy established maintain highest ethical standard ensure well-being everyone involved
CCRPs provides principal investigator certification to ensure accuracy and efficiency for form completion and compliance.