
Good Pharmacovigilance Practice

Good Pharmacovigilance Practice Modules: A Comprehensive Guide to EMA GVP Modules
In 2025, guaranteeing the secure and compelling utilize of medications remains vital, and following to great pharmacovigilance hones is significant. The European Medications Organization (EMA) has set up comprehensive rules to guarantee the secure observing of drugs all through their lifecycle. Our pharmacovigilance preparing program offers certification in these modules, recognized by IAOCR and right now attempted by over 6,000 students.
Compliant with universal guidelines, great pharmacovigilance hones require that companies or showcasing authorization holders (MAHs) actualize strong frameworks to screen pharmaceutical security, assemble information on potential dangers, and instantly report any suspected unfavorable responses. Fast distinguishing proof of security issues and suitable activity by MAHs are imperative.
Moreover, EMA's Hazard Administration Plans give extra direction on overseeing item dangers all through their lifecycle. These plans address chance minimization methodologies, benefit-risk appraisals, extra observing frameworks, and post-marketing studies.
It is basic for pharmaceutical companies to follow to great pharmacovigilance hones to guarantee quiet security and the opportune conveyance of quality solutions without undue dangers or delays. Taking after these rules makes a difference defend open wellbeing by empowering quick discovery of security signals some time recently genuine hurt happens.
Pharmacovigilance Systems
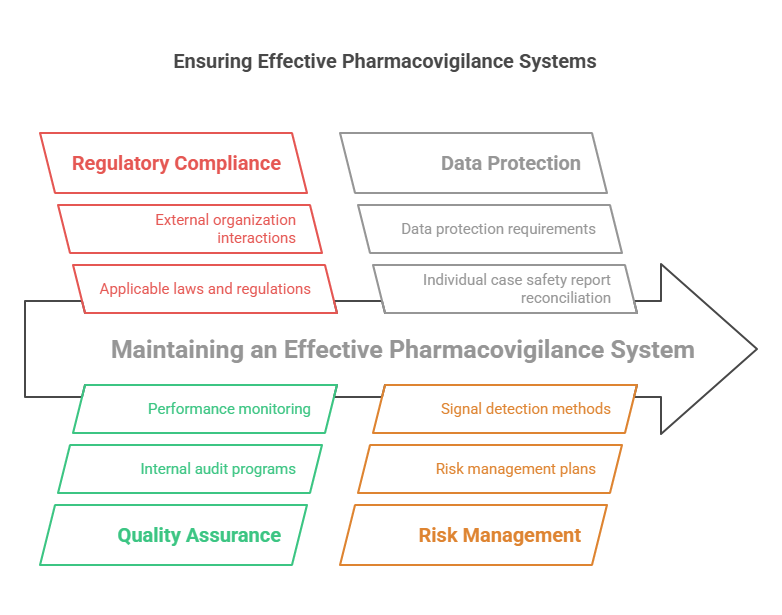
The GVP Module I – Pharmacovigilance Systems and Their Quality Systems outlines key principles, requirements and expectations for managing effective pharmacovigilance systems, as well as providing guidance on how to develop and maintain a quality system. The document provides a comprehensive overview of the regulatory framework and highlights the importance of an efficient, effective and compliant pharmacovigilance system. It includes information such as the definition of what constitutes a PV system, the role of relevant parties in setting up and operating such a system, expectations regarding PV processes and procedures, criteria for evaluating adequacy of PV systems, risk management activities, safety data exchange agreements and safety reporting procedures.
The document outlines the responsibilities associated with setting up a PV system and maintaining its quality assurance program. This includes ensuring that applicable laws or regulations are followed; obtaining the necessary resources (people, equipment); identifying appropriate roles for personnel involved in PV activities; developing policies, procedures and standards; monitoring performance; making sure that any changes to the system are properly validated/re-validated; implementing risk management plans; establishing an internal audit program; performing regular internal audits to ensure compliance with applicable laws or regulations; interacting with external organizations involved in safety surveillance activities. Additionally, it discusses topics such as data protection requirements, individual case safety report reconciliation processes, periodic safety update reports (PSURs), risk management plans (RMPs), signal detection methods and other related topics.
Overall this guideline is an essential reference tool for industry professionals responsible for setting up or maintaining pharmacovigilance systems. It provides a detailed description of all aspects relating to pharmacovigilance systems including those related to regulatory requirements, quality assurance programs and data protection measures. This makes it an ideal source of information for industry professionals looking to stay informed about best practices in this field.
Pharmacovigilance System Master File
The GVP Module II – Pharmacovigilance System Master File (Rev 2) is a comprehensive document that outlines the best practices for companies involved in the pharmaceutical industry to ensure the safe use of their products. The document covers topics such as the required contents of a pharmacovigilance system master file (PSMF), recommendations for setting up, running and maintaining a PSMF, as well as safety-related responsibilities for healthcare professionals and companies.
The GVP Module II provides clear guidance on what should be included in the PSMF, including elements such as organizational information, operational processes, safety data management and reporting, safety risk management and analysis, and communication processes. It also outlines good practices related to quality assurance such as validation of data entry systems and implementation of change control procedures. Moreover, it provides detailed instructions related to specific roles within a pharmacovigilance system such as medical advisors, clinical evaluation experts and signal detection staff.
In addition to providing recommendations on how to implement adequate pharmacovigilance systems, this guideline also includes discussion points on how companies can validate their own individual systems. This includes guidance on how to audit against established standards such as those outlined by the European Medicines Agency (EMA). Furthermore, it outlines requirements for drug development plans including preclinical studies, clinical trials, post-authorization studies and post-marketing surveillance programs.
Overall, this document is an invaluable resource for anyone involved in any aspect of drug safety or pharmacovigilance. It provides clear guidance about what constitutes an adequate pharmacovigilance system for both healthcare professionals and companies involved in the pharmaceutical industry. Its detailed descriptions make it easy to understand exactly what needs to be done from concept through implementation and operation of a PSMF. Additionally its discussion points provide valuable insights into how existing systems may be evaluated or improved upon if needed.
Pharmacovigilance Inspections
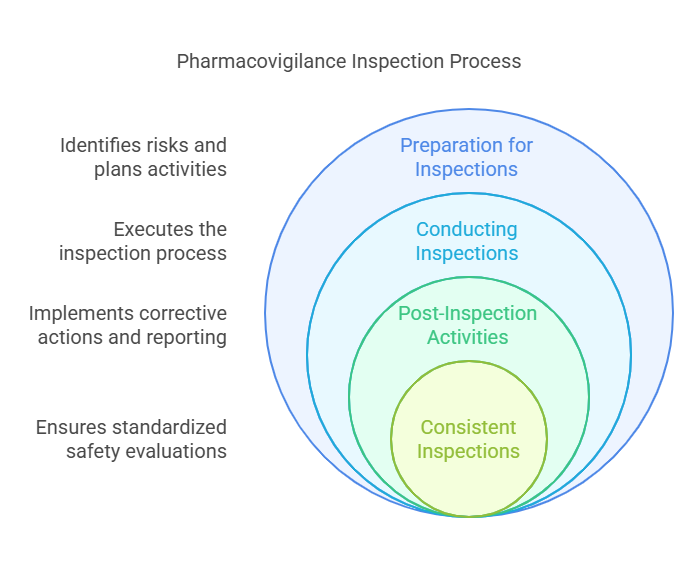
The GVP Module III – Pharmacovigilance Inspections (Rev 1) provides an overview of the inspection process and methodology used in pharmacovigilance. This document is intended to standardize the approach taken by authorities when carrying out inspections as part of their pharmacovigilance activities. The document starts by providing an overview of the objectives and scope of inspections, as well as a list of key elements that should be assessed during such inspections. It then goes on to outline the organization and conduct of inspections, including the roles and responsibilities of all those involved, before concluding with a discussion of post-inspection activities.
The document contains information on how to prepare for an inspection, including identifying risks, developing a detailed plan and appointing appropriately qualified inspectors. It also covers topics such as evidence gathering, reporting requirements, defining corrective and preventive actions (CAPA), handling disagreements between authorities and documentation requirements. The document also includes guidance on how to manage conflicts of interest during inspections, assess data integrity issues in clinical studies or establish a dialogue between authorities and inspected companies.
Overall, this guideline provides comprehensive information about conducting pharmacovigilance inspections. It sets out detailed instructions for all stages of such inspections – from preparing for them to taking corrective actions afterwards – helping ensure that these activities are carried out in a consistent manner across different countries. As such, this guideline is likely to be beneficial for both authorities responsible for managing safety concerns related to medicines and inspected companies which must comply with relevant regulations.
Pharmacovigilance Audits
The GVP Module IV provides guidance for conducting pharmacovigilance audits. This document is intended to provide an overview of the essential elements of a robust quality system and auditor qualification, planning and preparation for the audit, conduct of the audit, closure and follow-up activities, and reporting.
The main objectives of conducting pharmacovigilance auditing are to ensure that the pharmacovigilance system meets applicable regulatory requirements and industry standards, while also promoting continuous improvement in safety management. The document outlines the expectations for planning and preparing for an audit including scope, criteria, documents to be reviewed, personnel to be interviewed and potential sources of evidence. It addresses important considerations such as effective communication with stakeholders during planning and performance of the audit.
The document also covers criteria to be used when selecting auditors to ensure objective assessments. Qualifications should include relevant knowledge within the area of pharmacovigilance as well as experience in conducting audits. Additionally, it specifies standards for verbal/written communications with all parties involved during an audit including respect for confidentiality/privacy requirements.
The document describes principles related to conducting the audit including appropriate documentation methods such as notes from witness interviews or observation forms. Guidelines are provided regarding evidence gathering techniques such as sample size determination, selection of subjects or records to review, duration of observations and additional topics related to ensuring effective data collection techniques are employed when necessary.
Additionally, this guideline outlines requirements for properly closing an audit including findings discussions with all parties involved followed by appropriate action plans that address any non-conformities found during the process. This action plan should aim at remediation of deficiencies found during either corrective or preventative actions if necessary/justified as well as a timeline for completion/follow-up actions on actions taken. Lastly, it describes expectations related to reporting post-audit activities which should include written reports addressing nonconformities found along with recommendations on corrective actions taken or additional preventive measures needed in order to ensure compliance with GVP guidelines going forward.

Risk Management Systems
The Guideline on GVP Module V - Risk Management Systems, revised for 2020, is a comprehensive document provided by the European Medicines Agency that outlines a framework for risk management systems of pharmaceutical products. It provides detailed guidance for manufacturers and marketing authorization holders during their production and distribution of medicinal products in Europe. The guideline covers topics such as the safety assessment process, risk minimization activities, communication of safety information to healthcare professionals and patients, pharmacovigilance audit procedures and training requirements.
The main aim of GVP Module V is to ensure that the risks associated with medicinal products are managed effectively throughout their lifecycle. This is achieved through an effective risk management system (RMS). To this end, the GVP sets out five core principles that should be adhered to when designing and implementing an RMS: monitoring and evaluation of safety information; risk minimization strategies; communication of safety information; audit and inspection; and training requirements.
Each principle is then broken down into more detailed elements which manufacturers/marketing authorization holders should consider when designing their RMS. These include: establishing objectives for the RMS; setting up a robust infrastructure to monitor safety data; developing risk minimization plans; communicating safety information to stakeholders on a regular basis; conducting audits/inspections on a regular basis; ensuring staff are trained appropriately in pharmacovigilance practices; setting up reporting systems to enable timely alerts if any significant new risks are identified; assessing performance metrics regularly to ensure processes remain effective over time.
Individual Case Safety Reports
GVP Module VI provides guidance for companies and organizations in the collection and management of individual case safety reports (ICSRs) as well as their submission to regulatory authorities. The main purpose of this module is to ensure that pharmacovigilance activities are performed in a consistent and effective manner across the EU Member States, in order to protect public health, improve patient safety, and strengthen confidence in healthcare products.
The module covers topics such as process for ICSR collection, management, and submission process; risk management plan; responsibilities; quality control measures; data integrity requirements; monitoring of adverse events reporting systems; ICSR privacy considerations; electronic exchange of ICSRs between marketing authorization holders and national competent authorities; and post-marketing surveillance.
In addition to providing practical guidance on these topics, the module also outlines best practices for maintaining a comprehensive pharmacovigilance system. These include establishing an appropriate risk management plan for each authorized medicinal product, assigning roles and responsibilities appropriately, collecting timely ICSRs from all relevant sources (including spontaneous reports from healthcare professionals or patients), tracking safety signals on an ongoing basis, ensuring data integrity when exchanging ICSRs electronically with regulatory authorities, ensuring the security of personal data related to patients who report adverse reactions, and performing regular monitoring activities to assess compliance with pharmacovigilance obligations.
Overall, the Guideline on good pharmacovigilance practices (GVP) Module VI is a valuable resource for companies and organizations that seek to ensure that their pharmacovigilance operations are up-to-date with current regulations and standards. It provides useful information on how to develop an effective ICSR collection, management, and submission process while also emphasizing best practices for maintaining a comprehensive pharmacovigilance system that is compliant with applicable laws.
Period Safety Update Reports
The GVP Module VII is designed to provide guidance for the development and submission of periodic safety update reports (PSURs). This document provides information on the regulatory aspects, content and format for PSURs, as well as best practices for preparing and submitting them in accordance with the applicable risk management plan.
The GVP Module VII outlines the process needed to assess drug safety data from various sources, including spontaneous reports, clinical trials, epidemiological studies and post-authorization safety studies. It emphasizes that periodic safety reviews should be conducted at least annually and whenever new data suggests it is necessary. The main objective of a PSUR is to provide an assessment of the benefit-risk balance of a medicinal product over a defined period of time.
In order to ensure that all relevant data is accurately captured and tracked, the GVP Module VII recommends that companies maintain a comprehensive database containing both adverse event and non-adverse event information related to their products. This information should include any relevant clinical trial results or other relevant safety issues identified during pharmacovigilance activities. Additionally, the document outlines methods for evaluating reported events in order to identify potential safety signals.
Overall, Guideline on good pharmacovigilance practices (GVP) Module VII provides detailed guidance regarding the development and submission of periodic safety update reports (PSURs). It outlines processes for capturing, tracking, evaluating and assessing drug safety data from various sources. Furthermore, it discusses strategies for analyzing this data in order to identify potential safety signals which help inform regulatory decision making about a particular pharmaceutical product's risk-benefit balance over time.
Post Authorization Safety Studies
The GVP Module VIII provides detailed guidance on the post-authorisation safety studies (PASS). This document is designed to help drug manufacturing companies, regulatory agencies and other stakeholders understand their respective roles and responsibilities in designing and conducting PASS.
The document outlines the principles of good pharmacovigilance practice and incorporates several international standards including those from the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). The document also highlights the potential benefit of using available data sources such as Electronic Health Records and administrative healthcare databases, as well as patient registries.
In addition to general requirements related to PASS design, GVP Module VIII outlines specific requirements regarding patient eligibility criteria, study protocol adherence, sample size calculation and analysis, ethics/informed consent requirements, as well as specific reporting requirements.
The document contains clear instructions on how sponsors should prepare a detailed risk management plan (RMP) with respect to PASS. This includes outlining different types of safety monitoring procedures that should be conducted during a study. Furthermore, it provides guidance on providing adequate training to ensure appropriate execution of the RMP. Moreover, it outlines the importance of appropriately packaging and labelling test articles used in clinical trials to minimize any potential risks or harm associated with them.
Overall, GVP Module VIII provides comprehensive guidance for all stakeholders involved in Post-Authorisation Safety Studies by outlining clear roles and responsibilities as well assessing potential risks associated with these studies. It serves as an essential reference guide for manufacturers who wish to design effective PASS protocols that adhere to international standards in order to ensure patient safety while at the same time promoting innovation within the industry.

Signal Management
The GVP Module IX on Signal Management is a comprehensive set of guidelines which provides the framework for the appropriate management of signals and safety issues related to medicines. It outlines the process for detecting and evaluating potential safety issues and risks associated with medicinal products, as well as providing guidance on when additional investigations should be considered, and how to respond when safety signals are identified. The document focusses on areas such as risk minimisation plans, benefit-risk assessments, laboratory tests, product recalls and market withdrawals.
The GVP Module IX begins by outlining the definitions and concepts associated with signal management. These include definitions of what a signal is, how a signal should be classified, when it is appropriate to consider further action and how to differentiate between pharmacovigilance activities and regulatory actions. This section also provides guidance on data sources which can be used to identify signals, including both spontaneous reports from healthcare professionals or consumers as well as epidemiological studies.
The next section details specific aspects of signal management such as risk minimisation activities, benefit-risk assessments, laboratory investigations and product recalls or market withdrawals. It outlines key points such as: planning risk minimisation strategies in advance; monitoring effectiveness; assessing the benefit-risk balance at regular intervals; conducting laboratory tests which are relevant to safety issues; recalling or withdrawing products where necessary; ensuring availability of up-to-date information about risks associated with medicines; considering other types of regulatory action where appropriate; maintaining records of all decisions made related to signal management; and reporting/publishing new information regarding any changes in risk assessment/benefit-risk balance.
Finally, GVP Module IX provides detailed guidance on post-marketing surveillance activities which should be conducted following implementation of any risk minimisation plans. This includes setting up systems for monitoring changes in safety profile after introduction into use in humans or in the environment, implementing quality control processes for data capture & analysis, garnering collaboration from stakeholders (e.g healthcare professionals & customer feedback), sharing data with other organisations/authorities where appropriate ,and implementing communication plans so that stakeholders are kept informed of any changes in risk assessment/benefit-risk balance due to new evidence becoming available over time.
Additional Drug Safety Monitoring
The GVP Module X on Additional Monitoring offers a comprehensive guide to the principles, methods and processes of additional monitoring in the field of pharmacovigilance. This document outlines the purpose, rationale and requirements of additional monitoring activities as well as providing practical guidance for its implementation. The module is divided into five sections: Introduction; Overview; Objectives; Policies and Procedures; and Resources and Tools. In addition, it provides detailed best practice recommendations for each of these subject areas.
The Introduction section offers an overview of pharmacovigilance as well as outlining the structure and purpose of GVP Module X on Additional Monitoring. It also provides definitions for key terms related to this area such as safety surveillance, signal detection, signal assessment, signal evaluation, risk management plan (RMP), post-marketing commitment (PMC) etc.
The Overview section provides a general overview of additional monitoring including an explanation of its objectives, purpose and importance in the management of drug safety risks. It goes on to discuss how additional monitoring data can be used by authorities to make informed decisions regarding marketing authorization or changes to an authorized product's RMP. The section also looks at how regulators can assess the adequacy of existing safety information and consider whether further data collection should be undertaken through additional monitoring activities.
The Objectives section outlines in detail the objectives to be fulfilled when undertaking additional monitoring activities such as obtaining further safety information about a marketed product or conducting ongoing risk-benefit assessments necessary for regulatory decision making about medicine availability or changes to an authorized product's RMP. It also discusses how appropriate target populations can be identified in order maximize benefit from the activity while minimizing risk from potential harms caused by inappropriate use or misuse of medicines.
The Policies and Procedures section explains in detail what should be included when developing procedures for implementing additional monitoring activities such as setting objectives for data collection, deciding target populations for data collection, identifying relevant sources of information (including electronic health records) etc. It also covers legal considerations such as patient consent requirements when collecting personal data through registries or other sources outside hospital settings etc., which are important when planning any form of clinical trial activity that uses anonymized patient data collected retrospectively from various sources (e.g., primary care centers).
Finally, the Resources and Tools section suggests some practical tools that may help with developing appropriate procedures when implementing additional monitoring activities such as questionnaires that could be used to collect patient reported outcome measures (PROMs) etc. In addition it outlines relevant international frameworks/agreements which must be taken into account when collecting global safety data sets through international registry networks such as those developed through ICH-GCP partnerships between different countries/regions across Europe or North America etc..
Overall this Guideline on Good Pharmacovigilance Practices Module X Additional Monitoring is an essential resource for anyone involved with designing or implementing pharmacovigiance systems since it provides comprehensive best practice advice that will help ensure safe use/distribution/monitoring of medicines worldwide
Pharmacovigilance Safety Communication
The GVP Module XV Safety Communication was developed to provide guidance to pharmaceutical companies and other healthcare stakeholders involved in the management of medicinal products. This document contains detailed instructions on how to effectively communicate risk related information about medicinal products.
The guidelines are designed to ensure that such communication is consistent, timely and accurate. It also highlights the importance of making sure that both healthcare professionals and patients have access to sufficient information so that they can make informed decisions about the medicine they are taking.
The document outlines a number of principles for effective safety communication, including: ensuring that all risk related information is identified and included in the communication; providing clear, accurate, up-to-date information; understanding who needs to be informed; responding quickly to questions raised by healthcare professionals; and making sure that patients have access to appropriate support after receiving information.
The guideline also sets out various requirements for monitoring and assessing the effectiveness of safety communications, including evaluating the impact of risk minimisation measures, collecting feedback from health professionals and consumers following safety communications, conducting surveys among healthcare professionals and monitoring changes in prescribing behaviour. The documentation also provides advice on dealing with adverse events associated with medicines as well as what steps should be taken when product recalls or withdrawals occur.
Overall, this Guideline on good pharmacovigilance practices (GVP) Module XV Safety Communication provides a comprehensive overview of best practices relating to safety communications concerning medicinal products. It provides specific advice on how pharmaceutical companies should communicate risk-related information about their medicines, as well as how to monitor the effectiveness of such communication. The guidance is invaluable for all stakeholders involved in managing medicinal products so that they can ensure patient safety is maintained at all times.
Conclusion
As we continue to navigate the complexities of drug safety in 2025, the role of Good Pharmacovigilance Practices (GVP) has never been more crucial. Each GVP module designed by the European Medicines Agency provides a comprehensive framework essential for maintaining the highest standards of drug safety and efficacy. For professionals looking to deepen their expertise or stay updated with the latest practices, our CCRPS offers a range of certification programs that not only meet these standards but also prepare you to excel in this critical field.
Want to understand good pharmacovigilance practice modules through examples, video lectures, and quizzes all while receiving The IAOCR internationally recognized certificate available for PV officers? Consider enrolling in CCRPS Pharmacovigilance certification.