
Top 100 CRA Certification Exam Questions with Complete Answers
Passing the CRA certification exam requires more than knowing definitions—it demands applying GCP, safety rules, and site protocols in real-world scenarios. Practicing with exam-style questions is one of the most effective ways to build that skillset and pass on your first attempt.
This guide includes 100 top CRA certification questions with complete, regulation-aware answers, grouped by key exam domains: GCP, ethics, protocol oversight, adverse events, monitoring, tools, and trial phases. Each answer is structured like the exam—clear, concise, and scenario-based. You’ll go beyond theory and start thinking like a certified CRA. If you’re just starting out, begin with foundational content like What Is Good Clinical Practice (GCP)?. From there, work your way up to complex monitoring dilemmas and documentation reviews.

Questions on Good Clinical Practice (GCP) and Ethics
1. What is the purpose of ICH-GCP in clinical research?
ICH-GCP sets a global ethical and scientific standard for clinical trials. It protects human subjects and ensures credible data collection.
Read: What is Good Clinical Practice (GCP)?
2. Who holds primary responsibility for GCP compliance during a trial?
The sponsor is ultimately accountable. However, investigators and CRAs ensure daily site-level adherence through monitoring and documentation.
3. What role does the Institutional Review Board (IRB) or Ethics Committee play?
They review protocols, informed consent documents, and trial amendments to protect participant rights and safety.
Explore: Understanding Institutional Review Boards (IRBs)
4. What are considered essential documents under GCP?
Documents such as the trial protocol, CRFs, informed consent forms, investigator brochures, and monitoring visit logs are essential to demonstrate compliance.
5. When is re-consent from a participant required?
Whenever there’s a significant protocol change, updated risk information, or a new consent form version.
6. What’s a common informed consent error seen during audits?
Using an outdated or unapproved consent form version, or obtaining consent after initiating study procedures.
See: Informed Consent – What Every Researcher Must Know
7. How should protocol amendments be handled at the site level?
They must be approved by the IRB before implementation, unless immediate changes are required to eliminate safety hazards.
8. What does a CRA check when reviewing consent documentation?
Proper signatures, accurate dating, version control, and assurance that no study procedure occurred before consent.
9. What should a CRA do when GCP violations are observed at a site?
Document the issue, notify the sponsor or CRO immediately, and ensure follow-up corrective action is tracked.
10. What is source data verification (SDV) under GCP?
It is the process of comparing trial data entered into the CRF with the original medical records or source notes.
11. Can a legally authorized representative (LAR) provide consent?
Yes, when the participant lacks decision-making capacity and if permitted by law and approved by the IRB.
12. What’s the immediate CRA response to an unsigned consent form?
Ensure the participant stops all study activities, notify the PI, and escalate the issue for resolution and documentation.
13. What is the Declaration of Helsinki?
A set of ethical principles developed by the World Medical Association guiding research involving human subjects.
14. Are verbal consents allowed in clinical trials?
Only when IRB-approved and thoroughly documented, including a witness’s signature or recording as per protocol.
15. When should protocol deviations be reported to the IRB?
Any deviation affecting participant safety, rights, or data integrity must be reported promptly, typically within 5–15 days depending on IRB policy.
Key Questions on GCP and Research Ethics
This Q&A covers essential ethical and regulatory standards every CRA must master before site monitoring begins.
- Purpose of ICH-GCP: Global standard ensuring subject protection and reliable data. Learn more
- Primary GCP Responsibility: Sponsors are accountable; CRAs and investigators ensure day-to-day adherence.
- IRB/Ethics Committee Role: Reviews protocols and consent materials to safeguard participants. Explore IRB roles
- Essential Documents: Protocols, CRFs, consents, brochures, and monitoring logs prove compliance.
- When Re-Consent Is Needed: After protocol changes or updated risk information.
- Common Informed Consent Error: Using outdated versions or post-procedure signatures. See full guide
- Handling Protocol Amendments: IRB approval is required before changes unless safety is at risk.
- CRA Consent Review Duties: Check for correct dates, signatures, and version control.
- CRA Response to Violations: Document, notify sponsor/CRO, and ensure follow-up actions.
- Source Data Verification (SDV): Confirm CRF entries against original source records.
- Use of LAR for Consent: Allowed if permitted by law and IRB-approved for incapacitated subjects.
- Unsigned Consent Form: Halt all procedures, notify PI, escalate for documentation.
- Declaration of Helsinki: Ethical framework guiding human subject research globally.
- Verbal Consent Rules: Permitted only with IRB approval and proper documentation.
- Reporting Protocol Deviations: Must be reported promptly if affecting safety or data integrity.
Protocol, IRB & Regulatory Oversight Questions
16. What’s the difference between a protocol amendment and a protocol deviation?
A protocol amendment is a planned change approved by the IRB before implementation. A deviation is an unplanned departure from the approved protocol.
17. What must be included in a protocol deviation report?
Date, description of the deviation, subject ID (if applicable), impact on safety or data, and corrective/preventive actions.
18. Who is responsible for ensuring protocol compliance at the site level?
The Principal Investigator (PI), supported by site staff and monitored by the CRA.
19. When can a CRA permit protocol deviations?
Never. Only the IRB and sponsor can approve changes; CRAs must report and document any deviations found.
20. What are common CRA findings related to protocol non-compliance?
Early dosing, missing labs, unreported adverse events, or unapproved consent versions.
21. What is the CRA’s role in IRB communication?
CRAs confirm that IRB approvals, continuing reviews, and amendments are current and documented in the regulatory binder.
Learn more: Understanding Institutional Review Boards (IRBs)
22. How often must IRB continuing reviews occur?
At least annually, though some high-risk studies may require more frequent reviews.
23. What documents must be submitted to the IRB for multi-site trials?
Full protocol, investigator brochures, informed consent forms, recruitment materials, and PI credentials.
24. What’s the difference between a central and local IRB?
Central IRBs review for multiple sites nationally; local IRBs serve individual institutions and may have stricter requirements.
25. How should a CRA confirm IRB approval?
Verify the IRB approval letter includes protocol number, version, approval date, expiration date, and site-specific PI name.
26. What happens if IRB approval lapses during a trial?
All study activities must stop until re-approval is obtained—except actions required for participant safety.
27. What is a regulatory binder?
A site’s official storage of essential documents, including IRB approvals, protocol versions, investigator credentials, and monitoring logs.
28. What is the 1572 form?
The Statement of Investigator form submitted to the FDA, outlining PI responsibilities and qualifications for IND trials.
Explore: Clinical Trial Protocol – The Definitive Guide
29. What does the CRA check during document review?
Current IRB approvals, protocol versions, PI CVs, training logs, and signed 1572s—all must be filed and current.
30. What does the term “regulatory oversight” mean in CRA work?
It refers to ensuring that all trial activities comply with applicable regulations, protocol requirements, and ethical standards.
| Knowledge Area | Key CRA Responsibilities | Oversight/Compliance Detail |
|---|---|---|
| Protocol Amendments vs Deviations | Recognize deviations, report promptly, never approve changes | Amendments must be IRB-approved; deviations must be documented with impact and CAPA |
| IRB Communication & Documentation | Confirm current approvals, continuing reviews, and correspondence are filed | Check approval letters for site PI, protocol number, and expiration dates |
| IRB Types & Review Schedules | Understand differences between central and local IRBs | Ensure continuing reviews occur annually or more frequently as required |
| Document Review & Regulatory Binders | Verify accuracy and completeness of essential documents | Must include protocol versions, PI CVs, IRB letters, 1572s, training logs |
| Regulatory Oversight Principles | Ensure site activity adheres to protocol, ethics, and law | CRA enforces GCP standards and monitors for audit readiness at all times |
| FDA Form 1572 | Ensure accurate, signed 1572 is filed and reflects current PI details | Clinical Trial Protocol – The Definitive Guide |
Questions on Adverse Events, Safety Reporting, and Risk
31. What is the difference between AE, SAE, and SUSAR?
AE (Adverse Event): Any untoward medical occurrence.
SAE (Serious Adverse Event): Results in death, hospitalization, disability, or is life-threatening.
SUSAR (Suspected Unexpected Serious Adverse Reaction): An SAE that’s unexpected and possibly related to the investigational product.
More: Adverse Events – Identification & Management
32. Who is responsible for initial AE reporting at the site?
The site’s Principal Investigator. The CRA ensures it's reported and documented correctly.
33. What AE documentation should be reviewed during monitoring?
Source documents, AE logs, progress notes, and CRF entries for consistency and timely reporting.
34. What are CRA responsibilities in SAE follow-up?
Verify timelines, supporting documentation, and submission to IRB/sponsor as required. Ensure narratives are accurate.
35. What’s the regulatory deadline for reporting SAEs to sponsors?
SAEs must be reported immediately, typically within 24 hours of site awareness.
36. When must a SUSAR be reported to regulators?
Fatal/life-threatening SUSARs: within 7 days; others within 15 days, as per ICH-GCP guidelines.
37. What does “relatedness” refer to in AE reporting?
Whether the event is causally linked to the investigational product. Assessed by the investigator.
38. How does a CRA verify that AEs are reported on the CRF?
By comparing AE logs and source notes to ensure all events are entered accurately and consistently.
39. What defines an unreported AE?
An event documented in source files but missing from CRFs or AE logs.
40. What questions help assess AE seriousness during monitoring?
Did it require hospitalization? Was it life-threatening? Did it cause disability or require medical intervention?
41. How do CRAs ensure safety data integrity?
Check timeliness of reporting, completeness of narratives, consistency across forms, and adherence to protocol.
42. What is risk-based monitoring (RBM)?
A strategy where monitoring resources are focused on high-risk data and activities rather than 100% SDV.
43. What is a risk mitigation plan at the site level?
A plan to manage potential protocol risks—like delayed lab reports or high enrollment errors.
44. What does a safety visit include?
Reviewing AE/SAE documentation, checking reconciliation logs, ensuring correct version usage of safety forms.
45. When should an AE be followed up with additional queries?
When data is incomplete, unclear, or lacks clarity on severity, relatedness, or outcome.

Clinical Trial Monitoring & Site Visit Questions (46–65)
46. What’s the purpose of a Pre-Study Visit (PSV)?
To evaluate site feasibility, PI engagement, available facilities, and protocol comprehension prior to trial activation.
Read: Site Selection & Qualification Visits – Essential Guide for CRAs
47. What documents are verified during a PSV?
Investigator CVs, medical licenses, SOPs, IRB approvals, equipment logs, and the site’s delegation of authority.
48. What is Source Data Verification (SDV)?
Cross-checking CRFs with original documents like lab reports and medical notes to confirm data accuracy and GCP compliance.
49. What’s the CRA’s role during the Site Initiation Visit (SIV)?
Train staff on protocol procedures, AE/SAE documentation, CRF completion, ICH-GCP compliance, and site expectations.
50. What triggers an unscheduled monitoring visit?
Frequent protocol deviations, data inconsistency, enrollment irregularities, or sponsor-initiated audit preparations.
51. How often are monitoring visits conducted?
Typically every 4–8 weeks, depending on enrollment rate, protocol complexity, and sponsor monitoring strategy.
52. What’s included in an Interim Monitoring Visit (IMV)?
SDV review, IP reconciliation, AE documentation, training checks, and updated essential document reviews.
Read: CRA Essential Monitoring Techniques
53. How do you verify IP (Investigational Product) accountability?
Compare dispensing logs with shipment records, verify temperature logs, and reconcile returns versus dispensed doses.
54. What defines a protocol deviation?
Any unapproved divergence from trial procedures, eligibility criteria, or visit schedule defined in the protocol.
55. What actions follow detection of a major deviation?
Document it in the monitoring report, notify the PI and sponsor, initiate a CAPA, and report to the IRB.
56. How should visit findings be documented?
Summarize observations, issues, resolutions, and recommendations in a Monitoring Visit Report within 5–7 days.
57. What’s the purpose of a Follow-Up Letter?
To formally communicate findings, request corrective actions, define deadlines, and ensure site accountability.
58. What should be done before a Close-Out Visit (COV)?
Ensure query resolution, final IP return, document archival, regulatory binder completion, and database lock readiness.
59. What’s the CRA’s role in audit preparation?
Confirm protocol adherence, complete documentation, training compliance, and clean TMF for inspection readiness.
Read: Investigator Site Management Mastery
60. What’s a key CRA site performance metric?
Low deviation frequency, prompt data entry, accurate AE reporting, and timely resolution of sponsor queries.
61. What must be in the regulatory binder?
IRB approvals, PI CV/license, protocol/amendments, AE logs, IP logs, delegation logs, and communication records.
62. How do CRAs evaluate staff training?
Check protocol-specific training logs, confirm GCP certifications, and quiz staff on critical trial procedures.
63. What are monitoring red flags?
Delayed AE reports, missing source documents, CRF inconsistencies, unsigned ICFs, or absent temperature logs.
64. What are CRA duties in remote monitoring?
Access eTMF/EDC systems, review uploaded documents, flag discrepancies, and coordinate follow-up virtually.
65. What’s typically on a site visit agenda?
Team huddle, SDV, AE review, IP audit, document verification, and discussion of action items with the PI.
Poll: Which Site Visit Phase Do You Feel Least Confident About?
CRA Tools, Acronyms, and Documentation Questions (66–75)
66. What is the purpose of the Case Report Form (CRF)?
The CRF captures all protocol-specified data for each participant. It ensures standardization, supports regulatory audits, and contributes to final study reporting.
67. What belongs in the Trial Master File (TMF)?
Key documents such as the protocol, IRB approvals, PI CVs, ICFs, monitoring reports, SAE logs, and delegation of authority logs must be maintained in the TMF throughout the trial.
68. What’s the difference between eCRF and paper CRF?
eCRFs are electronic and allow real-time data capture and query management. Paper CRFs require manual entry and are more prone to transcription errors.
Read: Top 100 Acronyms in Clinical Research Explained Clearly
69. What is ePRO and how is it used?
Electronic Patient-Reported Outcomes (ePRO) systems allow patients to self-report symptoms and quality-of-life data, usually through tablets or web apps, enhancing accuracy and compliance.
Read: Directory of Electronic Patient-Reported Outcomes (ePRO) Tools – 2025 Edition
70. What is a deviation log?
A running list maintained at the site to track all protocol deviations, including date, subject ID, type of deviation, and resolution status.
71. What is the purpose of a Delegation of Authority Log?
This document lists staff authorized to perform study tasks, with their start and end dates, signatures, and roles—critical for audit readiness.
72. What are common CRA acronyms every monitor must know?
Examples include: SDV (Source Data Verification), TMF (Trial Master File), ICF (Informed Consent Form), IP (Investigational Product), AE/SAE (Adverse Events/Serious Adverse Events).
Read: Top 20 Clinical Trial Monitoring Terms Every CRA Should Know
73. What does EDC stand for?
Electronic Data Capture—a centralized platform for entering and managing clinical data in real time.
74. What is the Investigator Site File (ISF)?
The ISF contains essential site-level documents mirroring the sponsor’s TMF, maintained by the PI or study coordinator.
75. How are clinical acronyms tested in CRA exams?
Expect acronym-based multiple-choice questions and scenario prompts that assess understanding of key documentation and data workflows.
Essential CRA Tools, Acronyms & Documentation Practices
CRA certification exams and real-world monitoring both require fluency in core documentation and system tools. From CRFs to EDC platforms, understanding how each component functions ensures audit readiness and data integrity across clinical sites.
- CRFs (Case Report Forms): Capture all protocol-specific participant data and support regulatory submissions.
- TMF & ISF: Central and site-level repositories for protocols, ICFs, approvals, CVs, and monitoring reports.
- eCRF vs Paper CRF: Electronic forms reduce errors and enable real-time query resolution; paper forms require manual oversight.
- ePRO Systems: Let patients self-report outcomes digitally. Explore top ePRO tools
- Delegation Logs: Identify who’s authorized to perform study tasks, including dates, roles, and signatures.
- Deviation Logs: Track all protocol violations with corrective actions noted.
- Key Acronyms: SDV, TMF, ICF, IP, AE, SAE, EDC—memorize and apply each in monitoring reports and site documentation. Review CRA terms
- Exam Tip: CRA exams test acronyms not just by name—but through their use in documentation review and compliance scenarios.
Questions by Clinical Trial Phase (76–90)
76. What is the focus of Phase I trials?
Phase I trials primarily assess safety, tolerability, dosage range, and pharmacokinetics. They’re usually conducted on small groups of healthy volunteers.
Read: Phase I Clinical Trials Explained
77. What makes Phase I units unique?
They operate in controlled environments with continuous monitoring, focusing on adverse reactions and drug metabolism.
78. What are Phase II trial objectives?
To evaluate drug efficacy in patients with the target condition, determine optimal dosing, and identify side effects.
Read: Phase II Clinical Trials Goals
79. How are endpoints selected in Phase II?
Endpoints are typically clinical improvements, biomarker changes, or measurable symptom relief, aligned with study objectives.
80. What is the ethical concern in placebo-controlled Phase II trials?
Ensuring equipoise—researchers must believe there’s genuine uncertainty about which treatment is better, and that patients aren't denied standard care.
81. What distinguishes Phase III trials?
They involve large patient groups across multiple centers to confirm efficacy, monitor adverse effects, and support regulatory approval.
Read: Phase III Clinical Trials – Definitive Guide
82. How are recruitment targets managed in Phase III?
CRAs monitor site performance metrics, enrollment logs, and screen failure rates, and escalate lagging sites to sponsors.
83. What documentation burden increases in Phase III?
Due to scale, Phase III demands higher volume of source documents, regulatory updates, IP tracking, and monitoring visit frequency.
84. What is Phase IV?
Post-marketing studies assessing long-term safety, real-world effectiveness, and rare adverse events.
Read: Phase IV Clinical Trials Clearly Defined
85. What does long-term follow-up in Phase IV require?
Robust retention strategies, patient contact tracking, and remote or hybrid monitoring to ensure compliance over time.
86. How are CRAs involved in Phase IV studies?
They review patient diaries, ensure pharmacovigilance reporting, and facilitate site support for long-duration protocols.
87. Are endpoints in Phase IV regulatory-driven?
Not always. Many endpoints are observational, economic, or patient-reported for real-world evidence gathering.
88. What is the role of Real-World Data (RWD) in Phase IV?
CRAs help collect RWD from EHRs, registries, or mobile apps to support pharmacoeconomic analyses and safety surveillance.
89. What are the key risks in Phase IV?
Patient dropout, under-reporting of adverse events, and variability in practice standards across real-world sites.
90. How do CRAs handle site performance in Phase IV?
Through remote data reviews, KPI monitoring, and consistent communication to sustain quality and data completeness.
| Clinical Trial Phase | Core Objectives | CRA Responsibilities |
|---|---|---|
| Phase I | Assess safety, tolerability, dosage, and pharmacokinetics in healthy volunteers. | Monitor for AEs, verify dosing logs, oversee lab data collection, and ensure consent accuracy. |
| Phase II | Evaluate efficacy, refine dosage, and identify short-term side effects in patients with the condition. | Verify endpoint alignment with protocol, monitor consent/version control, and manage AE documentation. |
| Phase III | Confirm efficacy and safety across large, multicenter populations for regulatory approval. | Track site metrics, manage IP and source documents, escalate deviations, and prepare for audits. |
| Phase IV | Post-marketing studies to collect long-term safety, real-world effectiveness, and RWD. | Review ePROs, ensure pharmacovigilance, oversee retention, and monitor KPIs remotely. |
Real-World Scenario-Based Questions (91–100)
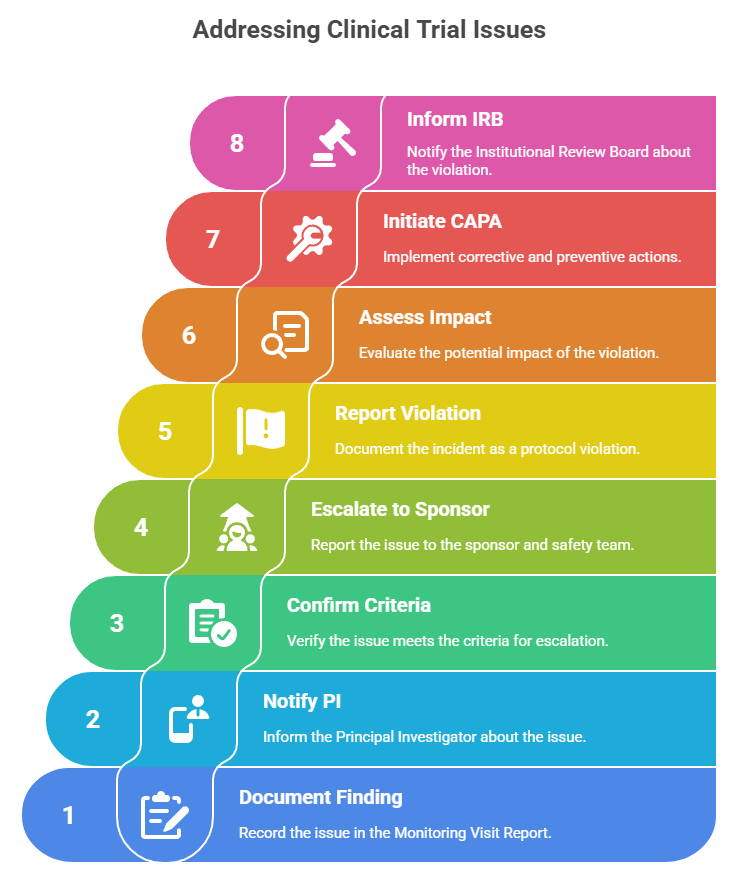
91. You find an undocumented SAE during a monitoring visit. What’s your first step?
Immediately document the finding in your Monitoring Visit Report, notify the PI, confirm the SAE criteria, and escalate to the sponsor and safety team per SOPs.
92. A site staff member shares unblinded treatment data with a patient. What’s your response?
Report it as a protocol violation and potential GCP breach. Document the incident, assess impact, initiate CAPA, and inform the sponsor and IRB.
93. The PI is unresponsive to repeated data query follow-ups. What should a CRA do?
Escalate through formal sponsor channels, document communication attempts, and request PI availability during the next monitoring visit or consider retraining.
94. During remote monitoring, you identify missing AE documentation. Next steps?
Flag the issue in the EDC, send a follow-up to the site, and request source documentation upload for verification and reconciliation.
95. You observe incorrect informed consent version used at enrollment. What do you do?
Notify the PI and IRB. Document the deviation, ensure re-consenting with correct form, and verify other patients aren’t affected.
96. A patient withdraws but their IP wasn’t returned. How should you proceed?
Initiate an IP reconciliation check, inform the PI and sponsor, and confirm drug storage or destruction per protocol.
97. A site uploads lab data with inconsistent units. What’s your role?
Query the discrepancy, ensure consistent unit conversions, and confirm alignment with protocol-defined reference ranges.
98. You discover multiple staff signatures missing on the delegation log. Action?
Request immediate updates, verify staff training records, and flag for sponsor review during audit prep.
99. Site staff report frequent power outages affecting data entry. What’s your mitigation plan?
Document the issue, assess risk to data integrity, suggest backup power or offline CRF options, and notify sponsor.
100. You detect recurring deviations in one protocol section. What now?
Initiate root cause analysis, suggest staff retraining, escalate if systemic, and monitor closely during next visits.
Final Thoughts: How to Use These CRA Exam Questions to Pass First Time
Practicing with real-world CRA certification questions is the single most effective way to build confidence and speed. This guide goes beyond memorization—it mirrors what you'll encounter on the actual CRA exam: regulatory logic, time-sensitive decisions, and documentation-heavy scenarios.
Make these questions part of your study rotation. Focus on understanding why each answer is correct, especially where GCP, protocol adherence, and monitoring decisions intersect. Use timing strategies, answer elimination, and regulatory rationale to improve not just your scores—but your career readiness.
To go further, enroll in the CCRPS CRA Certification Program, which offers expert-written materials, case-based quizzes, and mentoring aligned with these exact exam standards.
Frequently Asked Questions
-
The best approach is to use a structured plan that balances regulatory reading, concept application, and question practice. Start with foundational content like ICH-GCP, clinical trial phases, and safety reporting. Then move into scenario-based questions that test your ability to apply these principles. Include timed practice exams to simulate the actual testing environment. Focus on the “why” behind correct answers. Reinforce weak areas with repetition and notes. Group study, flashcards, and discussions can further solidify knowledge. Studying consistently over a few weeks—not cramming—is the key to building long-term retention and real-world exam confidence.
-
On average, preparation takes between four to eight weeks depending on your background in clinical research. If you're already working in the field or have familiarity with regulatory concepts, you may need less time. Those new to clinical trials, site visits, or safety reporting may require closer to ten or twelve weeks to be fully confident. Effective prep involves a blend of structured lessons, mock exams, and applied practice questions. Dedicate at least 10–15 hours a week and gradually ramp up in the final two weeks. Spaced repetition and timed review sessions also improve long-term success.
-
CRA exam questions are heavily weighted toward real-world application rather than pure memorization. You’re tested on how to interpret regulations, apply GCP standards, respond to protocol violations, and handle site management issues. Many questions are scenario-based and ask what a CRA should do in a specific situation involving monitoring visits, adverse events, or IRB communication. This format ensures you're ready for job tasks—not just textbook knowledge. To succeed, focus on how principles are implemented in practice and how your actions affect compliance, safety, and trial data quality across diverse clinical settings.
-
Yes, there are several types of free resources available to support your CRA exam preparation. These include blogs, regulatory guidance documents, glossary flashcards, and downloadable templates for visit checklists and deviation logs. Publicly available guidelines from regulatory bodies such as ICH, FDA, and EMA also serve as essential study tools. Forums and professional networks allow knowledge-sharing and study tips from peers. Some certification providers also offer free sample questions and exam prep outlines. However, while free resources can help, combining them with structured, curated content often provides a more complete and reliable study plan.
-
Failing the CRA exam isn’t the end—it's a diagnostic opportunity. Most certifying bodies allow retakes, often after a short waiting period. Use that time to assess what went wrong. Review the question types that challenged you, whether it was protocol comprehension, GCP application, or site documentation. Reflect on your test-taking strategies—did you run out of time, second-guess yourself, or miss key phrases in scenarios? After identifying gaps, restructure your study approach with more targeted practice, mock tests, and review cycles. With focused effort, most candidates succeed on their second attempt and emerge better prepared for real CRA work.