
GCP Compliance Strategies for Clinical Research Coordinators
Clinical Research Coordinators don’t fail GCP because they “don’t know the rules.” They fail because systems collapse under real-world pressure: rushed visits, conflicting instructions, missing signatures, unclear delegation, undocumented deviations, messy source, and last-minute monitoring panic. This guide gives you a field-tested GCP compliance operating system—the same kind of structure sponsors and monitors reward. You’ll get a 25+ control matrix you can implement immediately, plus practical strategies to reduce findings, protect subjects, and keep your site audit-ready—without becoming a paperwork robot.
1: GCP compliance for CRCs is a system, not a mindset
GCP is not “be ethical and keep good notes.” For a CRC, GCP compliance is repeatable control over risk—subject safety, data integrity, and documented oversight—across dozens of micro-processes that break easily when you’re understaffed. Your job is to design workflows that prevent preventable deviations (wrong visit window, missed labs, consent timing errors), keep your documentation defensible, and make it easy for oversight roles to verify what happened.
Start by anchoring your work to the parts sponsors actually evaluate:
Protocol execution: visit windows, required assessments, endpoint timing (see how endpoints get judged in primary vs secondary endpoints).
Documentation quality: source-to-EDC alignment, contemporaneous notes, controlled corrections (CRFs sit at the center—use CRF best practices).
Oversight & accountability: delegation, training, PI involvement, safety reporting (understand monitoring expectations from the CRA lens in CRA roles & skills).
Regulatory readiness: essential documents, version control, signature completeness (your fastest win is mastering document control—see managing regulatory documents).
If you want fewer findings, stop thinking “I should be more careful” and start thinking “Which control failed, and how do I make it hard to fail again?” That’s the CRC version of GCP.
| Process / Touchpoint | What “Good” Looks Like (GCP Standard) | Common Failure Mode | Evidence / Artifact to Prove It | CRC Strategy That Prevents Findings |
|---|---|---|---|---|
| Informed consent timing | Consent obtained before any study procedures | Vitals/labs collected “while waiting” | Signed ICF + note with timestamp + visit flow | Hard rule: “No consent, no procedure” checklist at front desk |
| ICF version control | Correct approved version used for that date | Old ICF pulled from drawer | IRB approval letter + version log + scanned ICF | Single-source binder + remove superseded versions immediately |
| Consent comprehension | Subject understands risks/rights | Rushed “sign here” approach | Consent discussion note + questions asked | Use a 3-question teach-back mini script in every consent |
| Re-consent triggers | Re-consent when protocol/ICF changes | Missed re-consent window | Change memo + subject contact note | “Re-consent roster” with due dates and escalation path |
| Eligibility confirmation | All I/E met, documented | “Assumed eligible” without proof | Eligibility worksheet + source labs | Two-step signoff: CRC verifies + PI reviews before randomization |
| Visit window adherence | Visits within protocol window | Scheduling drift | Visit schedule + source date + deviation assessment | Window calculator + reminders 10/5/2 days out |
| Protocol deviation handling | Identified, assessed, documented, reported per SOP | Deviation discovered at monitoring | Deviation log + CAPA + sponsor/IRB comms | Daily “exceptions huddle” (5 min) to capture deviations fast |
| Delegation of authority | Tasks delegated appropriately, current signatures | Staff performing tasks not delegated | DOA log + training records | DOA reviewed on every staffing change + monthly check |
| Training documentation | GCP + protocol training documented | “Trained verbally” with no record | Training log + certificates + attestation | Training packet required before system access / visit involvement |
| PI oversight | PI involvement evidenced and timely | PI “rubber-stamps” late | PI signoffs + notes + review logs | PI review calendar: weekly safety review + monthly data review |
| Source documentation | Contemporaneous, attributable, legible | Backfilled notes | Progress note with date/time/initials | Write visit note before subject leaves; use structured template |
| Corrections (ALCOA) | Single line, dated/initialed, reason if needed | Whiteout / overwriting | Corrected source pages | Correction SOP reminder at every workstation |
| EDC entry timeliness | Within sponsor expectations | Late entry causes query pile-up | EDC audit trail + data entry tracker | “48-hour data rule” + protected data-entry blocks |
| Query management | Queries answered accurately with source support | Guessing / inconsistency | Query response log + source reference | Use “source first” rule; escalate uncertainties to PI same day |
| CRF completion quality | Complete, consistent, conforms to definitions | Inconsistent units/terms | CRF guide + edit checks evidence | Maintain a “site data dictionary” for common fields |
| Randomization readiness | All prerequisites met before randomization | Randomize before full eligibility documentation | Eligibility checklist + randomization confirmation | “Gate” checklist required to access randomization system |
| Blinding protection | Blind maintained; procedures prevent unblinding | Careless notes reveal treatment | Blinding SOP + incident log (if any) | Red flag words list for notes; separate unblinded files |
| Investigational product accountability | Accurate receipt, storage, dispensing, return/destruction | Missing counts / temps out of range | IP logs + temp logs + reconciliation | Two-person checks for dispensing + weekly IP mini-audit |
| Temperature excursion handling | Excursions documented and assessed | “Not sure” what happened | Excursion form + sponsor guidance + CAPA | Excursion kit: form + contacts + quarantine labels |
| AE documentation | AEs captured, graded, assessed for relatedness | Only “serious” events documented | AE log + source note + follow-up | AE script at every visit: symptoms, meds, ER/urgent care |
| SAE reporting | SAEs reported within required timelines | Late notification to sponsor | SAE report + timestamped submission evidence | “SAE escalation tree” posted + on-call backup CRC |
| Concomitant meds | Captured accurately with start/stop, dose, reason | Missing OTCs/supplements | Conmed log + med reconciliation note | Ask by category: Rx, OTC, supplements, herbals every visit |
| Lab sample handling | Correct tubes, processing, shipping chain maintained | Mislabeled or late shipment | Shipping logs + lab requisitions + tracking | Pre-visit kit check + “label before draw” rule |
| Device calibration | Equipment calibrated per schedule | Expired calibration sticker | Calibration certificates + maintenance log | Monthly equipment walk-through checklist |
| Regulatory binder completeness | Essential docs current, filed, signed | Missing CVs, licenses, signatures | TMF/binder index + filing log | Weekly “binder hour” + versioning + signature chase list |
| Monitoring visit readiness | Source, binder, IP, logs ready | Scramble creates errors | Pre-monitor checklist + issue tracker | Run pre-monitor audit 7 days prior, not the night before |
| CAPA effectiveness | Corrective actions prevent recurrence | Same findings repeat | CAPA plan + follow-up evidence | Write CAPA tied to root cause + measurable proof step |
| Confidentiality / privacy | PHI protected; access controlled | Loose paperwork / shared logins | Access logs + privacy training | Clean desk policy + role-based access + shred bins |
2: Build a CRC “compliance engine” that survives real site pressure
The fastest path to GCP compliance isn’t “work harder.” It’s a simple engine:
Standardize the visit flow
Every visit should feel boring in the best way—same sequence, same checkpoints, same closeout. Add “hard stops” where errors happen: consent timing, eligibility, visit window, IP dispensing, lab handling. When your flow is consistent, you reduce the kind of downstream chaos that turns into CRF inconsistencies (tighten CRF thinking with CRF types & best practices).Document in a way that makes monitoring easy
Monitors don’t “find” issues because they’re looking to punish you. They find issues because your documentation fails to prove intent, timing, and oversight. Think like a CRA: if you were monitoring your own site, what would you need to verify? That’s why reading expectations from the monitor side matters (see CRA roles, skills & sponsor expectations).Create one place where “truth lives”
GCP failures explode when information is scattered: separate trackers, emails, sticky notes, and half-updated spreadsheets. Your “single source of truth” can be a simple master tracker (screening, visit schedule, deviations, AEs, query aging). If you’re not sure what belongs where, use the organizing principles from managing regulatory documents for CRCs and keep all essential oversight artifacts controlled.Reduce data integrity risk at the point of capture
Data errors are cheaper to prevent than to correct. Treat endpoints like fragile assets—timing, methods, and documentation must match protocol intent (calibrate your mindset with primary vs secondary endpoints). If your study has complex measures, make the logic simple for staff: what must be done, when, how, and what proof is required.Make training operational, not ceremonial
Training isn’t “we did an investigator meeting.” Training is ensuring the person doing the task can do it correctly and document it properly under time pressure. If your site includes newer coordinators, consider building a structured learning path using CCRPS resources and continuing education directories like clinical research continuing education providers so training becomes a pipeline, not a scramble.
3: The five highest-risk GCP failure zones for CRCs (and how to lock each down)
1) Consent is a workflow, not a form
The risk isn’t just missing a signature. The risk is timing, version control, and comprehension. Your lock-down strategy:
Create a consent “kit” per protocol: current ICF, version log, short teach-back prompts, documentation template.
Use a same-day scanning rule so missing pages are caught immediately.
Document the discussion, not just the signature.
If your study has placebo elements, consent comprehension becomes even more critical—subjects must understand uncertainty and assignment structure (see how placebo designs work in placebo-controlled trials).
2) Deviations become findings when you hide them
Sites that try to “avoid deviations” often create worse outcomes: unreported issues discovered later, loss of trust, and repeated problems without CAPA. Instead:
Capture deviations daily (2-minute huddle).
Classify quickly: safety impact? endpoint impact? compliance impact?
Document root cause and prevention action.
A deviation often cascades into endpoint risk—especially when timing matters (again: endpoints clarified with examples). Your goal is to protect interpretability, not pretend perfection.
3) Data integrity breaks at source, not in EDC
If the source is weak, you will fight queries forever. Strong source documentation is:
Contemporaneous
Specific (who/what/when/why)
Consistent with protocol definitions (use your CRF guidance: CRF definition, types & best practices)
If your protocol includes complex procedures (randomization, blinding), source needs to clearly show gates were met and blind was protected (see randomization techniques and blinding types & importance).
4) Safety documentation gets “thin” under workload
CRC teams often focus on “serious” safety items and miss the quieter ones: persistent symptoms, medication changes, ER visits, new diagnoses. Build a safety capture routine that’s impossible to skip:
AE script every visit
Med reconciliation every visit
Clear PI review cadence
If you interface with PV teams or safety databases, you’ll also benefit from understanding safety vocabulary and workflows (see what is pharmacovigilance).
5) Oversight artifacts (DOA, training, binder) decay quietly
Your binder doesn’t break loudly—it rots in small ways: expired CVs, outdated licenses, missing signatures, uncontrolled versions. The fix is boring but powerful:
Weekly “binder hour”
Monthly DOA reconciliation
Training evidence required before role access
Use the structure in managing regulatory documents and calibrate expectations by reviewing what CRC responsibilities look like when done professionally (see CRC responsibilities & certification).
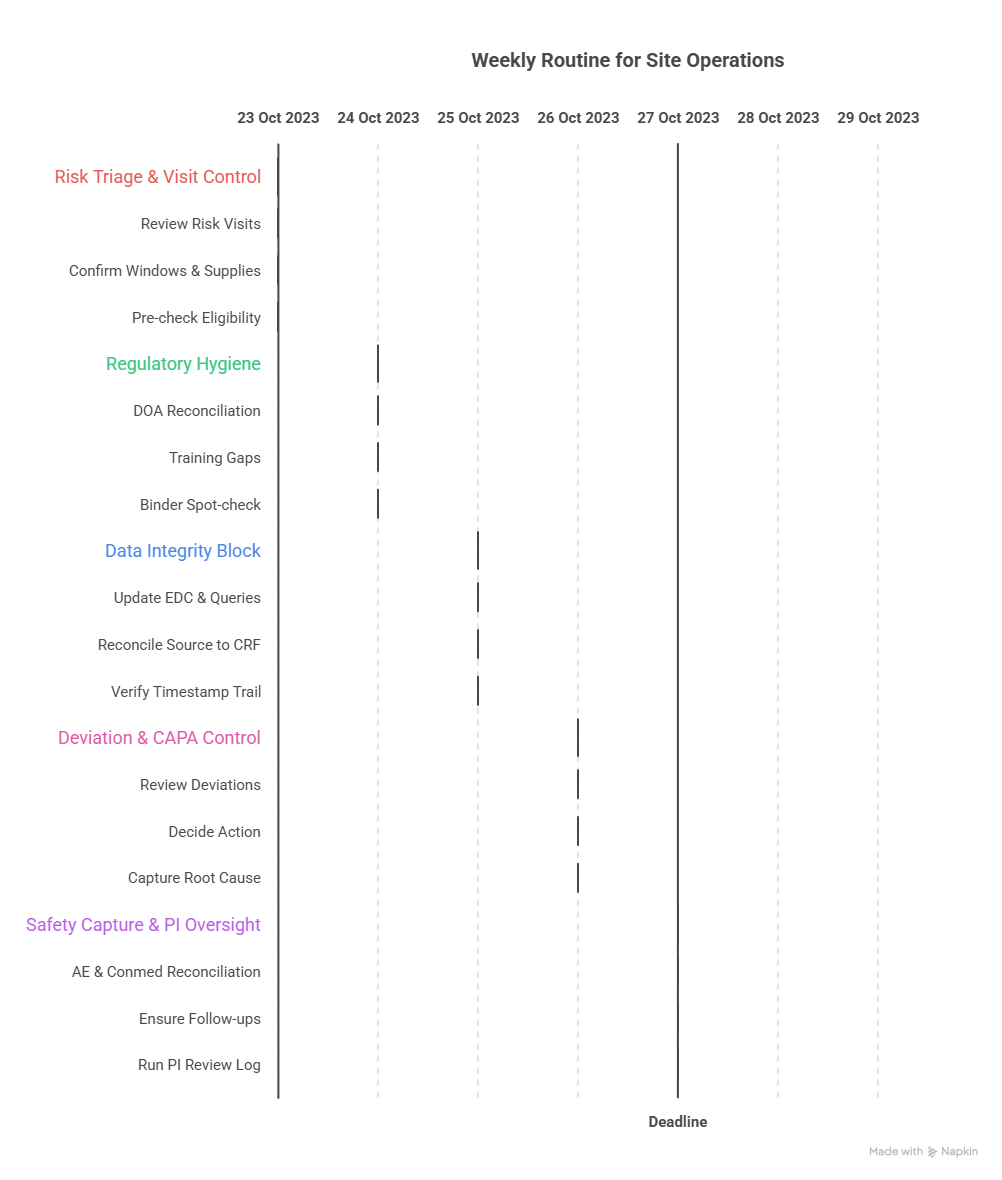
4: A practical weekly GCP compliance routine CRCs can actually sustain
Most sites don’t need a 40-page SOP overhaul. They need a weekly routine that prevents decay. Here’s a high-leverage cadence that stops problems before they become findings:
Monday: Risk triage + visit window control
Review the week’s visits and identify “risk visits” (screening/randomization, first dose, key endpoints).
Confirm windows and required assessments; pre-stage supplies and templates.
Pre-check eligibility gates for any randomization candidates (align with randomization techniques).
Tuesday: Regulatory and oversight hygiene (1 hour)
DOA reconciliation: who performed what tasks last week vs what’s delegated.
Training gaps: any new staff? new protocol memo? document it.
Binder spot-check: top 10 essential docs, signatures, version control (use regulatory documents guide for CRCs).
Wednesday: Data integrity block (protected time)
Update EDC for recent visits and attack your oldest queries first.
Reconcile source → CRF fields most likely to trigger edit checks (use CRF best practices).
If endpoints are timing-sensitive, verify the timestamp trail is defensible (reinforce with endpoints clarified).
Thursday: Deviation and CAPA control
Review deviations logged this week (even “minor” ones).
Decide: report? document? CAPA? PI review needed?
Capture root cause while memory is fresh. A late CAPA often reads like fiction.
Friday: Safety capture + PI oversight evidence
AE and conmed reconciliation from the week’s visits.
Ensure follow-ups are scheduled and documented.
Run a quick PI review log check; oversight is often judged by proof, not intent.
If you want to level this up faster, push structured upskilling into your routine using the CCRPS ecosystem—e.g., resources like clinical research continuing education providers or a broader credential comparison via certification providers directory.
5: How to “win” monitoring and audits without becoming a documentation machine
Monitors and auditors reward three things: predictability, transparency, and proof. You can deliver all three without drowning.
1) Make your documentation decision-based
Most weak notes describe events. Strong notes document decisions:
why an assessment happened late
what was done about it
who was informed
what prevention step was implemented
This reduces “mystery gaps” that trigger queries and follow-up letters. It also makes your deviations defensible instead of suspicious.
2) Treat complex trial design elements as compliance multipliers
If your trial uses blinding, randomization, placebo, and strict endpoints, each one multiplies compliance risk. Build guardrails:
Blinding guardrails: separate unblinded access, careful language in notes (see blinding types & importance).
Placebo clarity: ensure subject understanding and documentation of expectations (see placebo-controlled trials).
Randomization gates: do not randomize “because it’s late and everyone’s here” (see randomization techniques).
3) Prevent “query debt” like financial debt
Query backlogs create desperation—and desperation creates bad answers. Your best move is to keep query aging small:
answer queries in batches during protected blocks
document source properly so future queries drop
standardize tricky fields with a mini site data dictionary (paired with CRF best practices).
4) Use professional networking to solve operational bottlenecks
Sometimes compliance is blocked by operational reality: staffing, vendor delays, training gaps, unclear best practices. The best CRCs learn fast by tapping the right circles:
for peer process improvements, use clinical research networking groups & forums
for targeted discussions, consider best LinkedIn groups for clinical research
to reduce career friction while upgrading skills, see CRC responsibilities & certification and CRA career path expectations
5) Build credibility with “preventable deviation” posts internally
Your sponsor, PI, and monitor trust grows when you proactively share fixes:
“We had a window near-miss—here’s our new reminder workflow.”
“We saw repeated conmed omissions—here’s the new reconciliation script.”
This mirrors what high-performing sites do and aligns with the discipline in regulatory document management.
6: FAQs on GCP compliance strategies for CRCs
-
Pick the top three failure zones: consent workflow, source documentation, and oversight artifacts (DOA/training/binder). Then implement a weekly routine that produces proof. If you can defend your process like a monitor would evaluate it, findings drop quickly—especially when your documentation aligns tightly with CRF standards (use CRF best practices) and your regulatory binder is controlled (see managing regulatory documents).
-
Don’t write a story—write a decision trail: what happened, when discovered, subject impact, data/endpoint impact, who assessed it (PI), whether it was reported, and what prevention step stops recurrence. Tie your assessment to protocol intent and endpoint relevance (see endpoints clarified with examples) so the deviation is framed in scientific terms, not excuses.
-
The big ones: missing timestamps, vague notes (“patient okay”), undocumented PI oversight, uncontrolled corrections, and source that can’t prove eligibility gates before randomization. If you run randomization or blinded trials, documentation quality matters even more—study design increases the penalty of ambiguity (see randomization techniques and blinding explained).
-
Standardize. Use a consent kit, remove old versions, document comprehension with teach-back prompts, and adopt a “no consent, no procedures” hard stop. If your study includes placebo arms or complex risk tradeoffs, you must document comprehension more clearly because misunderstanding is more likely (see placebo-controlled trials).
-
You don’t win by heroic effort—you win by reducing complexity. Protect your data-entry blocks, run small weekly audits using the control matrix, and build training into your system using structured resources like continuing education providers and certification providers comparisons. You can also learn faster by using curated communities such as networking groups & forums and LinkedIn group directories.
-
Yes—because CRAs are paid to detect and manage the risks CRCs live inside. If you can show you built systems that reduce deviations, protect blinding, and keep documentation audit-ready, you’re demonstrating CRA instincts. Map your current work to monitor expectations using CRA roles & career path and reinforce your CRC fundamentals via CRC responsibilities & certification.