
Protocol Deviations: Definition, Examples & Corrective Actions
Protocol deviations are rarely “small paperwork issues.” In real trials, they are warning lights. A missed visit window can distort endpoint timing. An ineligible subject can compromise safety and data integrity. A missed lab can erase the only early signal that a participant was heading toward harm. Teams that treat deviations casually usually discover the real cost later: audit pain, rework, sponsor distrust, protocol amendment churn, and findings that could have been prevented with tighter execution.
That is why deviation mastery is a core clinical research skill, not a niche compliance topic. Whether you work in GCP compliance, clinical trial documentation, CRC responsibilities, CRA oversight, or audit readiness, the teams that handle deviations well protect subjects, preserve data, and build trust that stands up under inspection.
1. Protocol deviations: clear definition, why they matter, and what teams often get wrong
A protocol deviation is any instance in which study conduct departs from the IRB-approved, sponsor-approved, or regulatorily required protocol procedures, visit schedules, eligibility rules, treatment instructions, safety assessments, or other study requirements. That sounds straightforward, but the real-world problem is not the definition. The problem is inconsistent judgment. One site calls something minor. Another calls the same event major. One coordinator files a note-to-file and moves on. Another escalates immediately. That inconsistency is exactly where inspection risk begins.
The most useful way to understand deviations is to stop thinking of them as a paperwork category and start thinking of them as control failures. A deviation means the trial planned one thing and executed another. The key question is not whether the change was intentional. The key question is what the departure affected: subject safety, informed consent validity, eligibility integrity, endpoint reliability, investigational product accountability, or regulatory compliance. Teams that already understand informed consent procedures, clinical trial protocol management, managing regulatory documents, clinical trial auditing, and patient safety oversight usually detect deviation seriousness faster because they see the downstream consequences, not just the form entry.
Another common misunderstanding is assuming all deviations are equal. They are not. A subject arriving one day outside an allowed visit window may be manageable if it does not affect treatment timing, endpoint capture, or safety monitoring. Enrolling a subject who failed inclusion criteria is an entirely different level of risk. Missing a required ECG after dosing may expose the subject and damage the study’s ability to characterize safety. Using the wrong version of consent can undermine the legal and ethical basis of participation. This is why deviation systems must be rooted in risk, not bureaucracy.
A good deviation process answers five questions fast. What happened? Why did it happen? What was impacted? What must happen now to protect the participant and study? What will prevent recurrence? If your site cannot answer those five questions cleanly, your deviation management process is already too weak.
2. Types of protocol deviations and how to judge severity correctly
One of the most important skills in deviation management is classification. If your team misclassifies deviations, everything downstream gets weaker: reporting, trend analysis, CAPA design, retraining, sponsor confidence, and inspection defense. The goal is not to create fancy labels. The goal is to create repeatable risk judgment.
The broadest distinction is between deviations that primarily affect administrative compliance and those that affect safety, rights, welfare, scientific validity, or product accountability. Missing a noncritical filing deadline in an internal tracker is not the same as dosing before confirming a required pre-dose pregnancy test. Yet many sites still rely on vague phrases like “minor” and “major” without consistent written criteria. Better systems define seriousness around impact domains: subject protection, consent validity, eligibility integrity, dosing integrity, safety surveillance, endpoint integrity, and regulatory reporting obligations.
A second useful distinction is between one-off execution failures and repeat-pattern failures. A single delayed follow-up call matters. Five delayed follow-up calls in two months point to a process defect. Trending matters because regulators and sponsors care whether the site learned from prior errors. This is why deviation review should connect tightly to GCP compliance strategies for CRCs, GCP compliance essentials for CRAs, study documentation skills, research compliance and ethics, and regulatory responsibilities for principal investigators.
Teams also need to distinguish between preventable and condition-driven deviations. A subject hospitalized unexpectedly may miss a protocol visit for reasons outside site control. That still requires documentation and impact assessment, but the root cause is different from a missed visit caused by poor scheduling discipline. If the root cause is external, the corrective action may focus on contingency planning. If the root cause is internal, the corrective action must fix the broken process. Failure to make this distinction leads to fake CAPAs that sound official but change nothing.
A practical severity screen is to ask:
Did this deviation expose the participant to risk?
Did it affect consent, eligibility, dosing, or safety surveillance?
Did it threaten endpoint interpretability or data integrity?
Did it require sponsor, IRB, or regulator notification?
Could it recur tomorrow under the same workflow?
If the answer to several of those is yes, the deviation is not “small,” no matter how common it looks on paper.
3. Real protocol deviation examples and what strong teams do next
Examples are where deviation theory becomes operational. Teams often know the definition but freeze when something goes wrong in live study conduct. The difference between weak and strong sites is not whether deviations happen. It is how fast the team recognizes significance, contains risk, documents the event, and prevents recurrence.
Example 1: Subject enrolled despite failing exclusion criteria
A coordinator reads screening labs quickly, the PI assumes eligibility was confirmed, and the subject is enrolled. Two days later, a monitor identifies a lab value that should have excluded participation. This is a high-risk deviation because it affects eligibility, subject safety, and study validity. Strong teams do not start with blame. They start with containment. The PI evaluates immediate subject risk. The sponsor is notified with complete facts. The IRB is informed per requirements. The subject’s continuation is assessed case-by-case. Then the site reconstructs exactly where the failure occurred: incorrect worksheet design, lab threshold misunderstanding, lack of dual review, or rushed signoff.
This is where CRC protocol management, CRA documentation techniques, PI responsibilities, adverse event handling for PIs, and clinical trial auditing readiness all connect. The corrective action is usually not “staff reminded to be careful.” It is a redesigned eligibility worksheet, mandatory second-person verification, PI signoff rules, and retraining anchored to the actual failure point.
Example 2: Missed post-dose safety assessment
A subject is dosed, but the required one-hour post-dose ECG is missed because another urgent patient issue disrupted workflow. This is more than a scheduling slip. It is a safety surveillance deviation. Strong handling means documenting the exact timing, determining whether any symptoms occurred, informing the PI immediately, notifying the sponsor if required, and capturing whether the missing assessment affects subject management or data analysis. The site then reviews whether the problem came from poor staffing, missing role assignment, or a visit flow that overloaded one coordinator.
Teams already strong in adverse event reporting, drug safety reporting timelines, medical oversight, medical monitor review processes, and laboratory best practices know that missed assessments are often systems failures masquerading as isolated events.
Example 3: Wrong consent form signed
A subject signs an outdated consent form because the superseded version remained in an active folder. This exposes the study ethically and regulatorily because the subject may not have received the most current risk information. The right response is not to quietly replace the form. Strong teams preserve the record, report per policy, evaluate whether re-consent is needed immediately, and investigate the version-control failure. In many inspections, this kind of issue is viewed as a signal that document control is fragile more broadly.
Example 4: Repeat visit-window misses across subjects
If three subjects in one month miss the same visit window, you no longer have three isolated deviations. You have a process pattern. Strong sites trend the issue, ask whether window rules are unrealistic or poorly explained, assess reminder timing, and determine whether staffing coverage is driving the misses. This is where time management, clinical project planning, resource allocation, and stakeholder communication become deviation-prevention tools, not just management concepts.
What is your biggest protocol deviation pain point right now?
Choose one. The fastest fix depends on where your process breaks first.
4. Corrective actions that actually work instead of sounding good on paper
The phrase “corrective action” gets abused constantly in clinical research. Too many deviation logs contain weak responses like “staff re-educated” or “reminded to follow protocol.” Those are not always wrong, but by themselves they are usually too shallow. If the same deviation could happen again tomorrow under the same workflow, the correction was not real.
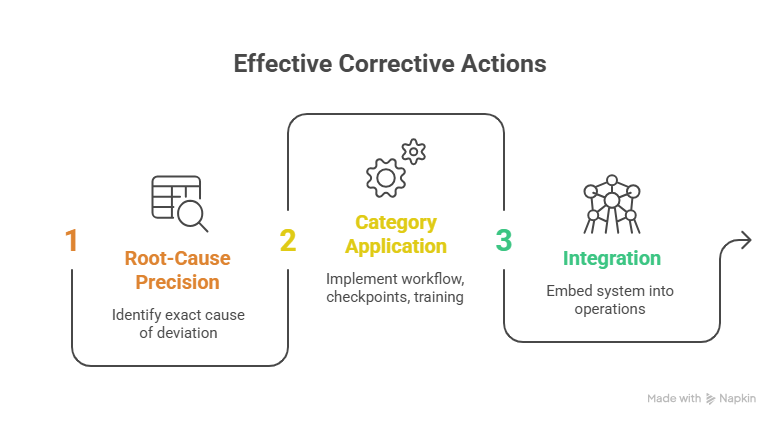
Strong corrective actions begin with root-cause precision. Not broad cause. Exact cause. Did the issue happen because the protocol was misunderstood, the visit workflow was overloaded, the source template missed a required field, the delegation log allowed untrained staff to act, the scheduling system did not flag window deadlines, or the PI review occurred too late? Unless you can identify the precise break point, your CAPA will be cosmetic.
Good corrective actions usually fall into five categories:
workflow redesign, decision checkpoints, document control improvements, training targeted to a real gap, and monitoring/trending reinforcement. For example, an eligibility deviation may require a locked screening worksheet, second reviewer signoff, mandatory PI confirmation before randomization, and a no-enrollment-until-complete rule. A missed PK sample may require a dedicated timekeeper, pre-labeled sample sequence board, role assignment before dosing, and fewer overlapping subject visits. A wrong-consent issue may require folder purge controls, color-coded current-version segregation, and eConsent access restrictions.
This is where managing regulatory documents, clinical trial documentation mastery, essential training under GCP, vendor management in trials, and risk management in clinical trials all become practical tools. A deviation system is strongest when it is integrated into operations, not trapped inside quality paperwork.
Another critical point: corrective actions should match the seriousness and recurrence risk of the deviation. Not every event requires a formal CAPA plan, but serious or repeated deviations do require deeper intervention and follow-up. The follow-up matters. A correction is not complete when the action is assigned. It is complete when the site can demonstrate the action changed performance.
5. How to prevent protocol deviations before they happen
Preventing deviations is less about telling staff to “be more careful” and more about making correct execution easier than incorrect execution. High-performing sites engineer the workflow so protocol compliance is the path of least resistance.
Start with protocol translation. Many protocols are dense, but deviations usually occur in a handful of high-risk zones: eligibility, consent, dosing, timed assessments, safety labs, prohibited medications, endpoint timing, specimen handling, and reporting timelines. Strong teams convert those into visit-specific tools. They use eligibility grids, pre-dose release checklists, timed procedure maps, source templates, consent version controls, prohibited-medication counseling sheets, and escalation trees. That approach aligns closely with case report form best practices, randomization techniques, blinding in clinical trials, data monitoring committee roles, and effective data collection and management.
Second, build redundancy into irreversible decisions. Randomization, dosing, eligibility confirmation, consent version verification, and SAE escalation should never depend on one rushed person operating from memory. Use second-person verification or hard-stop checkpoints where the risk justifies them. The best sites know exactly where a four-minute check can prevent a four-month remediation.
Third, trend near-misses, not just formal deviations. If staff repeatedly scramble to make a PK draw on time, that is a future deviation warning. If re-consent packets are repeatedly hard to locate, that is a future consent deviation warning. Near-miss intelligence is one of the most underused compliance tools in clinical operations.
Finally, create a culture where escalation is fast and non-defensive. Sites get hurt when coordinators hesitate to report an issue because they fear blame. A deviation caught early is usually manageable. A deviation hidden, softened, or discovered late becomes a credibility problem. Teams that combine GCP culture, ethical responsibility, PI leadership, CRC skill development, and CRA career discipline tend to create that kind of environment.
6. FAQs about protocol deviations
-
Organizations use the terms differently, but in many settings “violation” is reserved for more serious departures that affect subject safety, rights, welfare, or major data integrity elements. The smarter approach is not to obsess over terminology first. Focus on documented severity criteria, impact assessment, and required escalation.
-
Not always in the same way or on the same timeline. Reporting depends on local IRB policy, protocol requirements, sponsor expectations, and whether the event affects safety, rights, welfare, or study integrity. Teams should never guess. They should work from written reporting rules and escalation pathways.
-
No. Seriousness depends on what the visit contained and what was affected. Missing a visit that included critical safety monitoring, dosing, or endpoint collection may be serious. A small scheduling shift inside a clinically acceptable context may still be a deviation but with lower impact. Context matters more than labels.
-
Stabilize subject safety and data integrity first. Then document the facts while they are fresh, notify the right people quickly, and assess impact before jumping to conclusions. Weak sites rush to explanations. Strong sites secure facts, protect participants, and escalate appropriately.
-
Because the site addresses the symptom instead of the process defect. Repeat deviations usually mean workflow design is weak, training was generic, responsibility is unclear, or high-risk tasks rely too heavily on memory and heroics instead of structured controls.
-
A credible corrective action is specific, tied to the actual root cause, proportionate to risk, assigned to an owner, implemented on time, and shown to have worked. “Staff reminded” without proof of process change rarely convinces anyone.
-
Strong CRAs do more than identify issues. They spot patterns, compare site behavior against protocol risk points, test whether preventive tools exist, challenge weak CAPAs, and help sites move from reactive cleanup to predictable execution. That is where monitoring adds real value.