
Investigational New Drug (IND) Application: Clear Guide & Examples
An Investigational New Drug (IND) application is where promising science collides with regulatory reality. Teams often think the IND is just a submission milestone, but in practice it is a stress test of whether your preclinical package, manufacturing controls, clinical logic, safety monitoring, and operational discipline can withstand FDA scrutiny. A weak IND rarely fails because one section is missing. It fails because the story across toxicology, CMC, protocol design, and risk mitigation does not hold together.
For clinical research professionals, understanding IND mechanics is not optional. It sharpens how you interpret GCP guidelines mastery, manage clinical trial documentation under GCP, anticipate protocol deviations, strengthen regulatory submissions in pharmacovigilance, and support inspection-ready execution from day one.
1. What an IND application actually is and why it matters
An IND application is the formal request to the FDA to begin testing a new drug or biologic in humans. It is not merely a packet of forms. It is a risk-justification file that explains why the sponsor believes exposing people to the investigational product is reasonable at this stage. In effect, the IND answers four questions: What is the product, what evidence supports human exposure, how will it be manufactured consistently, and how will the proposed study protect subjects?
This matters because many teams misunderstand the true threshold. FDA is not asking whether the product is already proven effective. It is asking whether the available data support cautious clinical investigation. That means even brilliant mechanisms can be delayed if the nonclinical bridge is thin, the dose rationale is weak, or the manufacturing section cannot show adequate control. Professionals who already understand case report form best practices, primary vs secondary endpoints, randomization techniques, blinding in clinical trials, and data monitoring committee roles usually grasp faster why the IND is foundational to every later operational decision.
The IND also forces cross-functional honesty. Regulatory may want concise justification, CMC may want flexibility, nonclinical may want caveats, and clinicians may want broad eligibility. But FDA reviewers see the seams. If your investigator brochure implies one risk profile while your protocol eligibility ignores it, credibility drops. If your manufacturing description sounds stable while your release testing is still evolving, confidence drops. That is why strong IND preparation is inseparable from audit readiness for CRAs, regulatory document management for CRCs, patient safety oversight, and disciplined adverse event reporting.
A useful way to think about the IND is this: the application is the first place your development program must behave like a coherent clinical system rather than a collection of promising documents.
2. Core parts of an IND application, explained simply
At a high level, an IND contains three pillars: animal or laboratory data, manufacturing information, and the clinical study plan. But professionals gain leverage when they understand what each pillar is really trying to prove.
Nonclinical section: proving the first human exposure is justified
The nonclinical package supports the decision to dose humans. This includes pharmacology, toxicology, safety pharmacology, and any special studies that matter for the product class. The point is not volume. The point is relevance. A team can generate many studies and still fail to justify the proposed starting dose, escalation speed, target population, or monitoring schedule.
For example, a clean IND narrative connects animal toxicology findings to specific protocol controls. If preclinical work suggests hepatic liability, the protocol should reflect liver-related exclusion criteria, tighter lab monitoring, and explicit stopping rules. This is the same discipline behind strong adverse event handling for PIs, strong drug safety reporting, strong aggregate reports in pharmacovigilance, and thoughtful signal detection in pharmacovigilance.
CMC section: proving the product used in humans is controlled enough
Chemistry, Manufacturing, and Controls is where many otherwise strong programs get exposed. In early development, FDA does not expect commercial perfection. But it does expect that the investigational product can be produced with adequate identity, strength, quality, and purity for human use. Reviewers want to know what you are making, how you make it, how you test it, and how you know the material given to subjects is reasonably controlled.
This is why clinical teams should not treat CMC as “someone else’s section.” Stability limits affect site inventory. Reconstitution rules affect protocol feasibility. Release testing impacts startup timing. Excipient or formulation changes can trigger amendments. Anyone involved in clinical trial project planning, resource allocation, vendor management, and clinical trial budget oversight should understand that weak CMC ripples into every downstream milestone.
Clinical protocol section: proving the study itself is defensible
The protocol is the operational face of the IND. It tells FDA who will be enrolled, how dosing will occur, what safety measures exist, what endpoints matter, and how foreseeable risks are controlled. A credible protocol does not confuse complexity with rigor. It prioritizes safe execution, interpretable data, and decision-useful endpoints.
This is where smart teams build on fundamentals from informed consent procedures, clinical trial protocol management, effective stakeholder communication, GCP compliance for CRAs, and GCP compliance strategies for CRCs. An elegant Phase 1 protocol is not the one with the most procedures. It is the one that answers the right early questions without creating avoidable safety ambiguity.
3. IND examples: what strong versus weak submissions look like
The fastest way to understand IND quality is to compare realistic examples.
Example 1: Strong early oncology IND
A sponsor developing a targeted oncology agent submits a first-in-human IND. The nonclinical package includes relevant efficacy models, repeat-dose toxicology, genotoxicity work appropriate to the program, and a clear exposure margin analysis supporting the starting dose. The clinical protocol uses a conservative dose-escalation design, sentinel logic where appropriate, tightly defined DLT windows, and biomarker sampling tied to the mechanism. Eligibility excludes patients at predictable risk based on the tox profile. The investigator brochure, protocol, pharmacy manual, and safety management plan all tell the same story.
Why this works: the sponsor is not pretending uncertainty does not exist. Instead, the IND shows the sponsor understands where uncertainty lives and has built controls around it. That mindset mirrors strong risk management in clinical trials, strong patient safety oversight, strong clinical medical oversight, and disciplined medical monitor review processes.
Example 2: Weak neurology IND with a compelling mechanism but poor integration
A sponsor has promising animal data in a neurology indication and strong investor enthusiasm. But the IND is fragile. The proposed human dose is justified with optimistic extrapolation rather than conservative margin logic. Safety pharmacology findings are mentioned but not operationalized in the protocol. The CMC section describes a formulation that may change soon, yet the protocol and pharmacy instructions read as though product handling is already stable. Monitoring frequency is light, and stopping rules rely on vague phrases like “clinically significant abnormality.”
Why this struggles: the sponsor mistakes scientific excitement for regulatory readiness. FDA reviewers often react negatively when the package looks like it was assembled by silo instead of designed as a unified risk plan. This is exactly why professionals must care about study documentation management, research compliance and ethics, clinical research team leadership, and inspection preparation essentials.
Example 3: Strong sponsor-investigator IND in an academic setting
An investigator-initiated study uses an already marketed drug in a new indication. The sponsor-investigator does not have big-company infrastructure, but the IND is thoughtful. The rationale for repurposing is clear. Existing human safety experience is used intelligently, not lazily. The protocol narrows enrollment to reduce avoidable risk, consent language is transparent about uncertainty in the new indication, and safety reporting responsibilities are assigned concretely.
Why this works: resource limitations do not automatically create poor submissions. Lack of discipline does. Teams that master regulatory and ethical responsibilities for principal investigators, protocol development for PIs, clinical trial documentation techniques, and research assistant data management can often outperform larger but sloppier organizations.
What is your biggest IND application bottleneck right now?
Choose one. Your answer points to the fastest risk reduction move.
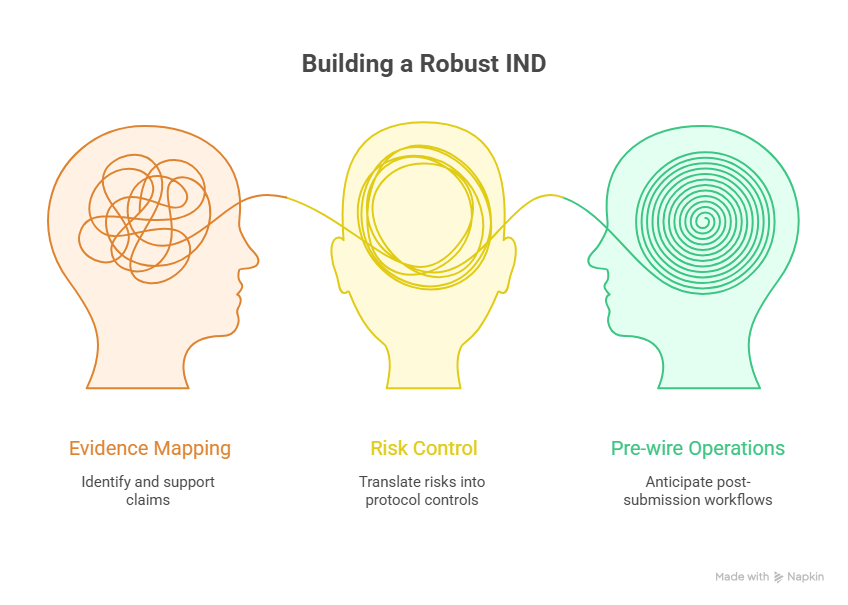
4. How to build an IND that survives real review pressure
The best IND teams do not begin with document templates. They begin with risk architecture. They ask: What are the top clinical unknowns? Which nonclinical findings truly matter for first-in-human risk? Which manufacturing variables could change interpretation of safety data? Which protocol decisions are essential, and which are just inherited habits?
Start with an evidence map, not a writing sprint
One practical method is to create an evidence map before authoring. List every major claim the IND makes, then identify the source data that support it. If a claim has no strong supporting evidence, either narrow the claim or generate the support. This prevents the common mistake of writing polished sections that cannot survive reviewer follow-up. Teams used to biostatistics in clinical trials, endpoint clarification, placebo-controlled trial logic, and DMC oversight usually do this naturally on the clinical side, but the same rigor must govern regulatory storytelling.
Translate every important risk into a protocol control
A critical IND principle is that known or plausible risks must show up as concrete study controls. That can mean eligibility exclusions, dose staggering, infusion management steps, ECG timing, lab thresholds, rescue medications, temporary hold criteria, or investigator escalation pathways. If a nonclinical signal never changes the protocol, reviewers may conclude the sponsor has not internalized its own data.
This is why professionals with hands-on experience in AE reporting for CRCs, CRA documentation discipline, GCP training requirements, and laboratory best practices often spot IND weaknesses earlier than people who only think in submission format.
Pre-wire post-submission operations before filing
Many sponsors act as though the IND ends at submission. In reality, the filing should already anticipate safety letter workflows, protocol amendment control, annual reporting obligations, site training, and document version management. A clean submission followed by chaotic activation still creates regulatory exposure. The teams that win are the ones that connect IND logic to startup, conduct, monitoring, reporting, and change control from the start.
5. Common IND mistakes that delay development and erode credibility
The most damaging IND errors are usually not dramatic. They are quiet credibility leaks.
One major mistake is forcing the clinical protocol to carry more ambition than the evidence supports. Teams under investor or internal timeline pressure often overload first-in-human studies with too many cohorts, too many exploratory endpoints, or overly broad eligibility. That may look efficient, but it increases protocol complexity before the safety foundation is secure. The result is slower enrollment, noisier interpretation, and more amendments. Professionals pursuing clinical trial manager career growth, clinical research project manager advancement, clinical operations leadership, and CRA career progression learn quickly that overdesign is often a hidden form of underthinking.
Another common mistake is treating CMC uncertainty as a drafting issue rather than a program risk. If formulation details are unstable, say so accurately and control the implications. Trying to make an immature product package sound fully settled often backfires. The same is true for toxicology caveats. Transparent limitations, paired with proportionate mitigation, earn more trust than inflated confidence.
A third mistake is weak cross-functional reconciliation. The nonclinical lead, regulatory writer, clinical scientist, safety physician, and CMC lead may all be individually competent yet still submit a package that disagrees with itself. These inconsistencies later become deviations, retraining, emergency clarifications, and monitor pain. Anyone who has lived through clinical trial auditing, regulatory submissions in pharmacovigilance, RMP planning, or medical monitor oversight knows the cost of unresolved ambiguity.
The final credibility killer is poor documentation of rationale. Teams often make smart decisions during meetings but fail to preserve the reasoning in a traceable, inspection-ready way. Months later, no one can explain why a dose changed, why a monitoring interval shifted, or why an exclusion criterion was added. That is how manageable scientific uncertainty turns into avoidable compliance risk.
6. FAQs about IND applications
-
An IND allows a sponsor to begin clinical investigation of an unapproved product or new use. An NDA is a marketing application submitted later to seek approval for commercial use. The IND is about justifying research exposure. The NDA is about proving the product should be marketed. Understanding this distinction helps professionals align early-stage risk logic with later-stage evidence expectations seen in clinical trial protocol management, GCP compliance essentials, clinical documentation, and stakeholder communication.
-
No. An IND does not mean the FDA endorses efficacy. It means the agency has not placed the proposed investigation on hold and the available information supports proceeding with clinical testing under the proposed controls. This is a safety-and-justification threshold, not proof of benefit.
-
A pharmaceutical company, biotech sponsor, academic institution, or sponsor-investigator can submit an IND. The key issue is not company size. It is whether the submitting party can meet regulatory responsibilities, including safety reporting, protocol control, investigator oversight, and recordkeeping.
-
Trouble often comes from weak starting dose rationale, poor translation of nonclinical signals into protocol safeguards, immature CMC controls, vague stopping rules, and contradictions across documents. In practice, the biggest danger is not one missing paragraph. It is a program that has not yet become operationally coherent.
-
Because IND logic shapes monitoring intensity, safety workflows, pharmacy handling, consent language, eligibility criteria, deviation risk, amendment frequency, and inspection readiness. If you understand the IND, you do not just follow the protocol better. You understand why the protocol exists in its current form.
-
Think like a reviewer trying to protect subjects while testing whether the sponsor truly understands its own product. Every important claim should have evidence. Every meaningful risk should have a control. Every cross-functional decision should remain traceable. That mindset improves not only submission quality, but the entire clinical system that follows.