
Site Monitoring Visits: Step-by-Step Guide for Coordinators
Site monitoring visits can either tighten a study’s execution or expose every weak habit a site has been hiding. For coordinators, the difference usually comes down to preparation, document control, source accuracy, IP discipline, and how confidently they can explain what happened at the site. A monitoring visit is never just a calendar event. It is a live stress test of your trial conduct, your GCP compliance, your protocol management, your regulatory document handling, and your readiness for the next audit, deviation review, or sponsor escalation.

1. What Coordinators Must Understand Before a Site Monitoring Visit Starts
A site monitoring visit is a structured review led by the CRA to verify that the study is being conducted according to the clinical trial protocol, ICH guidelines, sponsor requirements, and applicable regulations. For coordinators, that means the visit is not just about answering questions politely. It is about showing that your enrollment, visit execution, safety handling, source documentation, investigational product control, and regulatory binder maintenance can survive line-by-line review.
Many coordinators struggle with monitoring visits for one reason: they prepare reactively instead of operationally. They wait for the CRA’s confirmation email, then scramble to reconcile source notes, chase signatures, locate lab ranges, explain missed windows, and patch filing delays. That pattern creates avoidable findings. Strong coordinators build visit readiness into everyday workflow through disciplined study documentation management, tighter GCP compliance strategies for clinical research coordinators, cleaner informed consent procedures, and faster handling of protocol deviations.
A CRA usually evaluates whether participant rights were protected, whether reported data are backed by source, whether protocol-required procedures were done correctly, whether safety events were identified and escalated on time, whether essential documents are current, and whether investigational product accountability is accurate. That connects directly to your daily responsibilities as a coordinator in patient recruitment, informed consent best practices, adverse event reporting, and protocol execution.
The biggest mistake inexperienced coordinators make is treating the CRA as a critic instead of a control point. A strong monitor can help identify systemic risk early: recurring late data entry, incomplete delegation logs, source-template gaps, inconsistent eligibility verification, weak investigational product reconciliation, or safety narratives that do not match chart evidence. When a coordinator uses the visit well, the monitor becomes an early warning system before problems mature into audit readiness failures, sponsor distrust, or inspection exposure.
Another mistake is underestimating how interconnected site functions are. A missing signature is rarely just a missing signature. It can signal training gaps, poor workflow design, PI oversight weaknesses, or flawed delegation practices. A single late SAE report can point to weak triage, poor subject follow-up, confusion about the difference between AEs and SAEs, or an incomplete grasp of adverse events identification, reporting, and management. Monitoring visits reveal whether your site’s processes actually hold together under scrutiny.
That is why coordinators need a step-by-step playbook. You need to know what to review before the visit, what to organize during the visit, how to answer without overexplaining, how to document follow-up, and how to prevent the same findings from returning. This is where solid fundamentals from the clinical research coordinator role, practical research compliance and ethics mastery, and a clear understanding of good clinical practice stop being academic and start protecting your study.
Site Monitoring Visit Readiness Matrix for Coordinators (28 Critical Checks)
Use this before every on-site or remote monitoring visit to catch the issues sponsors escalate most often.
| # | Review Area | What the CRA Will Look For | Coordinator Action Before the Visit | Why It Matters |
|---|---|---|---|---|
| 1 | Visit schedule | Confirmed date, scope, and subjects selected for review | Request agenda, reviewed subjects, and expected access needs | Prevents day-of confusion and wasted monitor time |
| 2 | Enrollment log | Accurate screening, enrolled, screen fail, and withdrawal status | Reconcile master log with source and EDC | Detects hidden discrepancies across systems |
| 3 | Delegation log | Current roles, signatures, dates, and qualified staff assignments | Verify every study task maps to trained delegated staff | Weak delegation creates major compliance exposure |
| 4 | Training records | Protocol, amendment, safety, and system training completed | File missing certificates and sign training acknowledgments | Untrained staff involvement undermines data credibility |
| 5 | Regulatory binder | Current IRB approvals, licenses, CVs, and essential documents | Update expired documents and file pending correspondence | Shows site control and inspection readiness |
| 6 | IRB submissions | Amendments, continuing review, deviations, and safety notices filed properly | Match binder contents to submission tracker | Missing submissions create preventable findings |
| 7 | Informed consent versions | Correct approved version used for each subject and date | Cross-check consent dates against IRB approval periods | Consent errors can be study-critical |
| 8 | Consent process notes | Evidence of discussion, timing, signatures, and reconsent when needed | Review source note completeness for every enrolled subject | Rights protection must be documented, not assumed |
| 9 | Eligibility documentation | Each inclusion and exclusion criterion supported by source | Create criterion-by-criterion eligibility packets if needed | Enrollment ineligible subjects can threaten the study |
| 10 | Source documentation | Complete, attributable, legible, contemporaneous, and consistent notes | Resolve blanks, unsigned entries, and unclear corrections | Source weaknesses drive most monitoring friction |
| 11 | EDC timeliness | Data entry delays and unresolved queries | Enter backlog and prioritize aging open queries | Late data entry reduces sponsor confidence |
| 12 | Query management | Responses supported by source and not guess-based | Answer only after verifying chart evidence | Weak query responses create repeat findings |
| 13 | Protocol windows | Visits, labs, ECGs, and procedures done within allowed timing | Flag out-of-window events and prepare explanations | Window issues can become deviations |
| 14 | Protocol deviations | Identification, documentation, impact assessment, and reporting | Maintain deviation log with CAPA-ready detail | Untracked deviations signal loss of control |
| 15 | Adverse events | Complete capture, grading, attribution, outcome, and follow-up | Compare progress notes, hospital records, and AE log | Missed events create patient-safety and regulatory risk |
| 16 | Serious adverse events | Timely reporting with complete narrative and documents | Check awareness date, sponsor report date, and supporting records | Late SAE reporting is a major escalation trigger |
| 17 | Concomitant medications | Accurate start/stop dates, dose, reason, and consistency with AEs | Reconcile med list with clinic notes and subject interviews | Medication errors distort safety interpretation |
| 18 | Laboratory records | Normal ranges, flags, review signatures, and follow-up actions | File missing ranges and document PI review clearly | Lab handling often reveals oversight gaps |
| 19 | Investigational product receipt | Shipment records, temperature review, and receipt accuracy | Match delivery logs with pharmacy/site records | Receipt issues compromise accountability chain |
| 20 | IP accountability | Dispense, return, destruction, and balance reconciliation | Perform full count reconciliation before the visit | Drug accountability errors draw immediate attention |
| 21 | Temperature logs | Continuous storage records and excursion handling | Review gaps, min/max checks, and excursion follow-up | Storage failures affect product integrity |
| 22 | Subject status tracking | Active, withdrawn, completed, lost to follow-up status clarity | Update tracker and document final disposition | Prevents confusion during SDV and closeout planning |
| 23 | Communication log | Important sponsor, CRA, and IRB communications filed and traceable | Centralize key emails and action confirmations | Verbal memory is not a compliance system |
| 24 | CAPA follow-up | Prior findings addressed with evidence, not promises | Prepare proof of correction for previously cited issues | Repeat findings damage site credibility |
| 25 | PI oversight evidence | Documented review, sign-off, and medical decision involvement | Ensure PI review is visible in notes and logs | Oversight cannot be implied from title alone |
| 26 | Source-to-EDC consistency | Dates, values, outcomes, and statuses aligned | Spot-check high-risk forms before the visit | Reduces data clarification cycles |
| 27 | Access readiness | Workspace, EMR access, copier/scanner, and staff availability | Arrange logistics and protected document access early | Operational chaos weakens monitor confidence |
| 28 | Action tracker | Clear ownership and deadlines for issues found | Keep a live visit follow-up tracker during the review | Fast closure separates strong sites from reactive sites |
2. Pre-Visit Preparation: How Coordinators Should Get Ready Without Scrambling
The best monitoring visits are mostly won before the CRA arrives. Coordinators who do well use a standing readiness process, not a last-minute cleanup. Start by reviewing the visit confirmation, the monitor’s agenda, the subject list selected for source review, outstanding queries, follow-up items from the prior visit, and any new risks introduced by recent enrollments, protocol amendments, safety events, or staff turnover. That review should immediately connect with your regulatory document workflow, your GCP self-assessment mindset, and your site’s clinical trial start-up checklist discipline.
The first preparation layer is document control. Pull the current protocol, informed consent forms, delegation log, training records, screening and enrollment logs, subject status tracker, deviation log, AE and SAE trackers, investigational product records, temperature logs, lab certifications, normal ranges, IRB approvals, safety letters, and relevant email correspondence. Compare them against sponsor-required essential document expectations and the principles laid out in clinical trial documentation under GCP and managing study documentation essential RA skills. If you cannot locate something quickly, the CRA will notice that your site’s control over the trial is weaker than it should be.
The second layer is subject-level review. For every participant likely to be reviewed, confirm that consent was properly obtained, eligibility was documented before enrollment, visit procedures match the schedule of assessments, source notes support EDC entries, protocol deviations are logged where required, AEs are captured accurately, concomitant medications are consistent, and follow-up is documented. This is where coordinators should lean on a strong understanding of informed consent what every clinical researcher must know, phase-specific trial conduct, endpoint definitions, and clean case report form best practices.
The third layer is risk triage. Not every issue carries the same weight. Missing initials on a noncritical worksheet and enrolling a subject with undocumented eligibility are not remotely comparable. Coordinators need to identify what can become major sponsor concern fast: consent issues, late SAE reporting, repeated out-of-window procedures, recurring missed protocol assessments, undocumented PI oversight, unresolved investigational product discrepancies, and patterns of delayed source completion. Use the logic behind patient safety oversight in clinical trials, adverse event handling essential PI guidelines, and handling clinical trial audits to rank what needs immediate attention.
Preparation also means controlling access and time. The monitor should not spend the first ninety minutes waiting for EMR permissions, a conference room, pharmacy access, or a staff member who alone knows where the delegation log lives. Confirm workspace, chart access, scanner availability, pharmacy coordination, and PI availability for key questions. Strong coordination here reflects the same operational discipline needed in clinical trial resource allocation, stakeholder communication, and vendor management in clinical trials.
Finally, review prior monitoring letters. Sites lose credibility when the same findings appear visit after visit. If the last letter cited incomplete source, late query response, poor investigational product reconciliation, or weak deviation tracking, prepare evidence that the process changed. That might include revised source templates, a new AE reconciliation step, standing weekly query review, PI review checkpoints, or better training logs. Repeat findings tell sponsors that the site can acknowledge a problem without truly controlling it. Coordinators who break that cycle become indispensable.
3. During the Visit: How Coordinators Should Manage the Review Professionally and Precisely
When the visit begins, your job is to make review efficient without becoming defensive, vague, or overtalkative. Start with alignment. Confirm what the CRA wants to review first, which subjects are highest priority, whether pharmacy or lab review is needed, what outstanding action items remain, and whether there are sponsor-specific concerns tied to enrollment pace, deviations, data lag, or safety follow-up. This creates a working structure and mirrors the discipline expected in clinical research project planning, time management for CRCs, and strong coordinator role execution.
As the CRA performs source review, give documents in a controlled sequence. Do not bury critical records under unrelated paper. For each subject, the CRA usually needs consent, eligibility support, visit source, procedure documentation, AE follow-up, lab review, investigational product records if applicable, and evidence that EDC entries reflect source accurately. Your role is to retrieve quickly, answer exactly what was asked, and avoid improvising facts you have not verified. Clean monitor interactions depend on the same discipline taught by CRA monitoring techniques, site selection and qualification visit logic, and investigator site management mastery.
One high-value rule: distinguish between explanation and excuse. If a subject was out of window, explain the operational fact pattern, the subject impact, whether sponsor and IRB notification were required, and what preventive action now exists. If an SAE was reported late, identify the awareness date, the reporting date, the communication chain failure, and the correction made. If source notes were incomplete, describe whether the event occurred, what source exists elsewhere, how a compliant late entry will be handled, and how the site will prevent recurrence. Coordinators gain trust when they show command of deviation management, SAE reporting procedures, and drug safety reporting requirements.
Take live notes throughout the visit. Many coordinators rely on the CRA’s eventual letter and lose critical context. Keep a running tracker of reviewed subjects, issues identified, documents requested, commitments made, and deadlines discussed. That protects your site when several small findings are raised verbally across a long day. It also helps you distinguish between actual findings, clarifications in progress, and sponsor preferences that are not formal noncompliance.
Know when to escalate to the PI. A coordinator should not independently answer medically interpretive questions, justify eligibility decisions that require investigator judgment, or explain safety causality beyond what the PI documented unless the process clearly assigns that role and the site’s documentation supports it. Strong PI involvement is central to principal investigator responsibilities, regulatory and ethical responsibilities for PIs, and defensible patient safety oversight.
Professionalism during the visit also includes how you handle pressure. Some monitors are direct. Some are cautious. Some are excellent teachers. Some are simply trying to close a large visit load quickly. Your consistency matters more than their style. Fast retrieval, factual answers, visible organization, and calm acknowledgement of real issues create confidence. Defensive energy, disorganized binders, delayed access, and hand-wavy answers make every small issue feel bigger.
What is your biggest site monitoring visit stress point right now?
Choose one. The answer usually reveals the process gap hurting your study most.
4. The Highest-Risk Findings Coordinators Need to Prevent Every Time
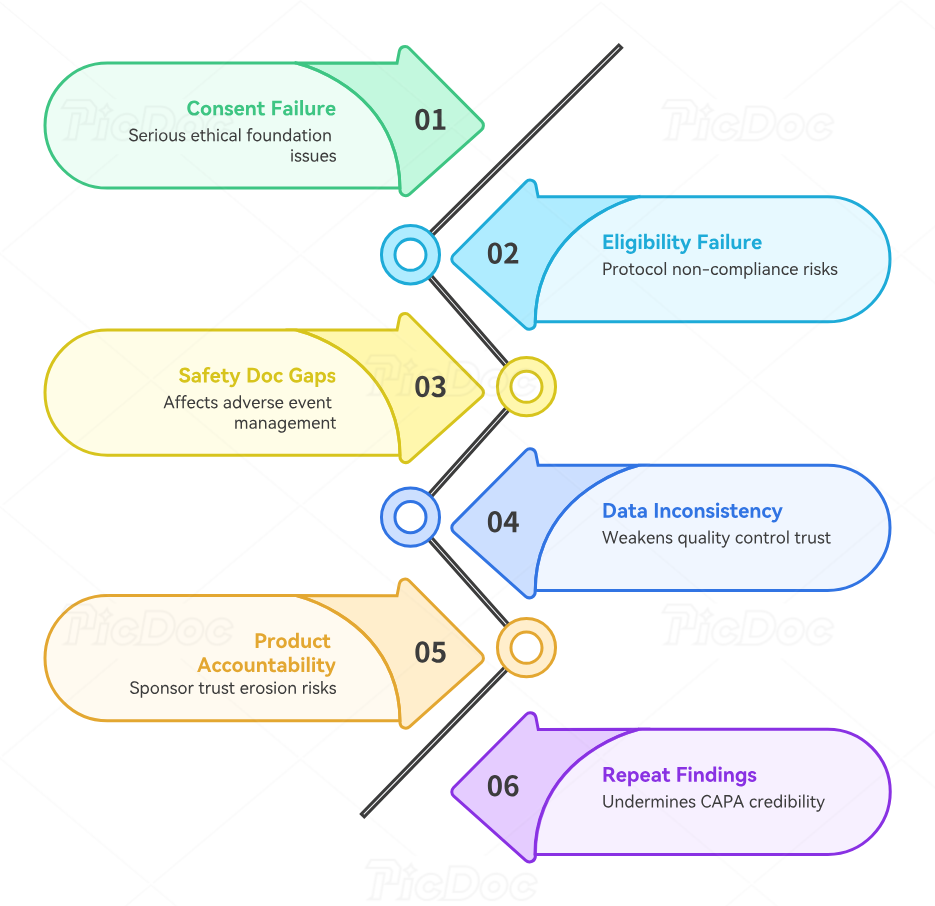
Some findings are annoying. Others can damage sponsor trust, trigger CAPAs, threaten enrollment continuity, or pull your site into deeper review. Coordinators need to know where the real danger sits. One of the most serious categories is informed consent failure. That includes using the wrong version, missing signatures or dates, consent obtained after procedures started, incomplete documentation of the consent discussion, and missed reconsents after an amendment. These problems hit the core ethical foundation explained in informed consent procedures, IRB roles and responsibilities, and research ethics resources.
Eligibility failures are next. A subject may look clinically appropriate and still be protocol-ineligible. Monitors will ask whether every inclusion and exclusion criterion was documented before enrollment and whether investigator review is visible. Coordinators get into trouble when they rely on memory, partial chart evidence, or verbal confirmation rather than criterion-linked documentation. This is where knowledge of protocol development and interpretation, primary versus secondary endpoints, and randomization basics becomes practical, not theoretical.
Safety documentation failures are another major danger zone. Coordinators often underestimate how much inconsistency appears between clinic notes, hospitalization records, AE logs, and EDC fields. The monitor may find an ER visit documented in source but absent from AE review, or a serious event reported to the sponsor without complete supporting narrative, or an outcome changed in the chart without corresponding follow-up in the database. These gaps directly affect adverse events management, serious adverse event procedures, pharmacovigilance fundamentals, and broader signal detection and management.
Source-to-EDC inconsistency is another repeat offender. Sponsors lose patience with sites that enter data late, answer queries weakly, or let values drift between systems. A blood pressure recorded one way in source and another in EDC may look minor alone, but across multiple visits it signals weak quality control. Coordinators should use habits similar to those required in clinical data management systems, EDC systems, and clean CRF practice: verify before entry, reconcile before response, and never answer queries from memory.
Investigational product accountability problems can escalate quickly. If counts do not reconcile, returns are undocumented, temperature logs have gaps, or shipment receipt records are sloppy, the sponsor’s concern moves beyond paperwork. They start questioning whether product was handled under controlled conditions and whether dosing records can be trusted. Coordinators need close partnership with pharmacy or designated drug staff and should understand the operational seriousness that mirrors clinical trial supply chain management, risk management in clinical trials, and GCP compliance essentials.
Finally, repeat findings are their own category of risk. A site that makes mistakes can recover. A site that repeats the same mistakes teaches the sponsor that corrective action exists only on paper. Coordinators need a live CAPA mindset: define the process failure, identify why it happened, assign a fix, retrain the relevant staff, verify that the new process is being used, and retain evidence. That is how monitoring visits stop being recurring ambushes and start becoming proof that the site is improving.
5. After the Visit: How Coordinators Should Close Findings and Build a Stronger System
The visit does not end when the CRA leaves. In many studies, the post-visit period is where strong coordinators separate themselves from overwhelmed ones. Start by organizing your own visit notes immediately. Confirm which issues were formal findings, which were pending clarification, which documents were requested, and which deadlines were discussed. Do this before memory blurs. That practice supports the same discipline needed in clinical trial auditing and inspection readiness, clinical trial documentation control, and study team leadership.
When the follow-up letter arrives, break each item into action parts. For example, “source documentation incomplete” is too vague to fix. You need to identify which forms, which visits, which staff, and what pattern caused the problem. Was the issue missing contemporaneous notes, undocumented PI review, weak AE follow-up detail, or no standardized template for unscheduled calls? The deeper the diagnosis, the more likely the correction will hold. Coordinators who rush into superficial fixes usually recreate the same finding at the next visit.
Assign ownership clearly. Not every action belongs to the coordinator alone. Some items require PI sign-off, pharmacy reconciliation, lab document retrieval, sponsor clarification, EDC updates, or retraining for sub-investigators and backup staff. Use an internal tracker with item, owner, due date, status, evidence, and prevention step. This approach mirrors the operational rigor expected in project management for clinical trials, budget and resource oversight, and effective stakeholder communication.
Your goal is not just closure. It is system strengthening. If consent corrections were needed, build a consent checklist and version-control habit. If deviations were underreported, add a weekly protocol variance review. If AEs were inconsistently captured, reconcile progress notes, subject calls, and hospitalization records at defined intervals. If query backlog was the problem, set a recurring review block. If delegation confusion caused errors, retrain staff and tighten who performs what. The best corrective actions improve workflow, not just paperwork.
Sites that perform well over time also normalize internal mini-reviews between monitoring visits. Pick a few active subjects each month and perform a focused internal review on consent, eligibility, AE capture, data timeliness, and IP accountability. This internal discipline draws on the same mindset behind interactive GCP self-assessment, essential training requirements under GCP guidelines, and managing protocol deviations. It is far easier to fix a problem you found yourself than one a sponsor escalates.
A final high-value habit is turning each monitoring visit into staff education. Review the most meaningful findings with your team, explain the operational consequence behind each one, and connect the lesson to patient safety, protocol integrity, data credibility, and sponsor trust. Teams get better when they understand not just what was wrong, but what risk it created. That is how coordinators become quality leaders rather than document chasers.
6. FAQs About Site Monitoring Visits for Clinical Research Coordinators
-
The coordinator’s main responsibility is to make the study transparent, reviewable, and defensible. That includes providing organized access to source documents, consent records, eligibility support, safety documentation, investigational product records, and essential regulatory materials while answering operational questions accurately. In practice, this means proving that the site’s daily conduct aligns with the clinical trial protocol, good clinical practice, and site-level CRC responsibilities.
-
Preparation should be continuous, but focused visit prep should begin as soon as the visit is scheduled. Waiting until one or two days before the visit almost guarantees rushed reconciliation, missing documents, and weak answers. Strong coordinators maintain readiness through ongoing regulatory document management, disciplined protocol management, and regular GCP compliance review.
-
The CRA usually reviews informed consent forms, source documents, eligibility support, visit documentation, AE and SAE records, concomitant medications, lab reports, investigational product accountability, temperature logs, delegation and training records, deviation logs, and key regulatory binder documents. Each of these ties into broader systems discussed in informed consent essentials, adverse event reporting techniques, clinical trial documentation, and CRA monitoring practice.
-
The most serious findings usually involve consent failures, undocumented eligibility, late SAE reporting, repeated protocol deviations, poor PI oversight evidence, major source-to-EDC inconsistencies, and investigational product accountability errors. These issues matter because they threaten participant protection, data integrity, or both. Coordinators should treat them with the urgency reflected in SAE procedures, protocol deviation management, patient safety oversight, and audit preparation essentials.
-
Answer honestly, then verify. Do not guess, fill silence with assumptions, or create a verbal explanation unsupported by records. A strong response is: “I want to verify that in the source or tracker before I answer.” That protects the study and your credibility. This habit supports clean documentation principles reinforced in research compliance and ethics mastery, GCP guidelines mastery, and clinical trial auditing readiness.
-
The best way is to convert each finding into a real process correction with ownership, training, verification, and evidence. Repeat findings happen when sites close paperwork without fixing workflow. Build a CAPA-style response: define the root cause, assign the fix, retrain the responsible staff, verify use of the new process, and review a sample of recent records to confirm improvement. This mindset aligns with interactive GCP self-assessment, training requirements under GCP, and stronger site management practice.