
Clinical Trial Safety Monitoring: Pharmacovigilance Best Practices
Clinical trial safety monitoring is where patient protection, regulatory credibility, and data integrity meet under real pressure. A trial can have a strong protocol, trained sites, and clean enrollment, but safety failure can damage everything quickly: participant trust, sponsor accountability, IRB confidence, and inspection readiness. Strong pharmacovigilance connects adverse event identification, serious adverse event reporting, drug safety timelines, medical monitor review, and GCP compliance into one disciplined safety system.
1. What Clinical Trial Safety Monitoring Must Actually Control
Clinical trial safety monitoring should detect, assess, document, escalate, and trend safety information before weak signals become participant harm or regulatory exposure. The work begins at the site, where coordinators collect symptoms, investigators assess clinical meaning, CRAs verify documentation, medical monitors review risk patterns, and pharmacovigilance teams process, code, reconcile, and report safety data. When this chain is weak, adverse events get underreported, SAEs miss timelines, source notes lack medical rationale, and sponsor teams discover safety gaps too late. Strong safety monitoring requires alignment across clinical research coordinator responsibilities, CRA monitoring techniques, principal investigator responsibilities, and pharmacovigilance fundamentals.
The first control point is complete adverse event capture. Sites should collect AEs actively, consistently, and neutrally rather than waiting for participants to volunteer problems. A coordinator who asks, “Any problems?” may miss fatigue, dizziness, rash, falls, medication changes, urgent care visits, and clinically significant labs. A stronger process uses visit-specific prompts, medication review, chart review, lab review, hospitalization checks, and device or diary review. That process protects AE reporting quality, patient safety oversight, source documentation, and case report form accuracy.
The second control point is seriousness assessment. A safety event becomes operationally dangerous when site staff delay escalation because they are uncertain whether hospitalization, life-threatening risk, disability, congenital anomaly, death, or medically important intervention applies. Pharmacovigilance best practice is to train sites on seriousness triggers before enrollment and reinforce that incomplete information should never delay initial reporting when SAE criteria may be met. This is where SAE procedures, drug safety reporting requirements, clinical trial medical oversight, and GCP training requirements become practical safeguards.
The third control point is clinical interpretation. Every AE and SAE needs severity, seriousness, causality, expectedness, action taken, outcome, and follow-up status handled with enough rigor to support decisions later. The PI should document medical judgment clearly, the medical monitor should challenge weak causality reasoning when needed, and the PV team should reconcile safety databases with EDC and source. Weak assessment language such as “probably unrelated” without rationale creates avoidable inspection pain. Strong assessment connects medical monitor responsibilities, principal investigator oversight, pharmacovigilance case processing, and clinical trial documentation under GCP.
| # | Safety Area | Common Failure Mode | Best-Practice Control | Proof That It Worked |
|---|---|---|---|---|
| 1 | AE collection | Participants mention symptoms casually, and staff fail to record them. | Use active questioning, chart review, lab review, and medication checks tied to AE identification. | Complete AE logs with onset, outcome, severity, and action taken. |
| 2 | SAE triage | Hospitalization or medically important events are escalated late. | Train site staff on seriousness criteria and same-day escalation rules. | SAE worksheet with reporting clock evidence. |
| 3 | Causality assessment | PI uses vague causality wording without medical rationale. | Require rationale linked to timing, disease state, concomitant medications, and study drug exposure. | PI-signed assessment with supporting source notes. |
| 4 | Expectedness review | Teams confuse known disease complications with expected product risks. | Compare events against protocol, IB, reference safety information, and medical review. | Expectedness decision documented in safety file. |
| 5 | Severity grading | Severity is confused with seriousness. | Train staff to grade intensity separately from seriousness criteria. | Consistent AE severity fields across source, EDC, and safety database. |
| 6 | Follow-up collection | Initial reports are submitted, then follow-up information disappears. | Track discharge summaries, labs, narratives, outcomes, and medication changes. | Follow-up tracker with due dates and final outcome status. |
| 7 | Source documentation | Safety data entered in EDC lacks source support. | Map AE fields to source templates and monitoring review expectations. | Source-to-CRF consistency confirmed during monitoring. |
| 8 | EDC-safety reconciliation | Safety database and EDC disagree on terms, dates, seriousness, or outcomes. | Run routine reconciliation between EDC, safety database, and site source. | Reconciliation report with resolved discrepancies. |
| 9 | Medical monitor review | Complex events are processed without medical challenge. | Escalate ambiguous causality, dose modifications, lab trends, and recurrent events to medical monitor review. | Documented medical monitor comments and actions. |
| 10 | Pregnancy reporting | Pregnancy exposure is treated as routine follow-up rather than special safety handling. | Define pregnancy reporting, follow-up, outcome collection, and infant follow-up requirements. | Pregnancy exposure tracker and outcome documentation. |
| 11 | AESI handling | Adverse events of special interest are missed because staff focus only on SAEs. | Train protocol-specific AESI triggers with examples and reporting routes. | AESI checklist in site safety workflow. |
| 12 | Lab abnormalities | Clinically significant labs are ignored or inconsistently documented. | Require investigator review, clinical significance assessment, and AE linkage when applicable. | Signed lab review with significance decision. |
| 13 | Concomitant medications | Medication changes reveal untreated AEs that were never recorded. | Review medication changes at each visit and cross-check with AE logs. | Medication-AE reconciliation notes. |
| 14 | Protocol deviations | Safety-related deviations are documented without root cause or CAPA. | Connect safety deviations to deviation corrective actions. | CAPA with effectiveness check. |
| 15 | Visit windows | Safety assessments happen outside required timing windows. | Use visit calculators, pre-visit reminders, and missed-assessment escalation rules. | Visit window tracker and deviation log. |
| 16 | Unblinding risk | Safety review exposes treatment assignment inappropriately. | Define blinded and unblinded safety workflows, access controls, and emergency code-break rules. | Blind maintenance documentation. |
| 17 | DSMB/DMC communication | Safety trends reach oversight committees late or in poor format. | Prepare clean listings, narratives, exposure data, and trend summaries for DMC review. | DMC package and decision log. |
| 18 | Signal detection | Teams process cases individually but miss emerging patterns. | Review frequency, seriousness, clustering, population risk, and exposure-adjusted trends. | Signal review minutes and action decisions. |
| 19 | Aggregate reporting | Periodic safety reports become administrative summaries with weak interpretation. | Use cumulative review, benefit-risk context, and medically meaningful trend analysis. | Aggregate report with documented conclusions. |
| 20 | Risk management | Known risks are listed but prevention actions are not operationalized. | Convert risks into monitoring checks, site training, participant instructions, and escalation rules. | Updated risk control plan. |
| 21 | Investigator training | Sites sign training logs but cannot apply safety procedures. | Use role-based safety scenarios during investigator meetings and refreshers. | Training records plus scenario completion proof. |
| 22 | CRA monitoring | CRAs verify AE fields but miss safety process failures. | Review source quality, PI review timing, follow-up status, and unresolved medical questions. | Monitoring report with safety action items. |
| 23 | Regulatory reporting | Expedited reports miss timelines because ownership is unclear. | Create a reporting responsibility map across site, sponsor, CRO, and PV vendor. | Reporting tracker with submission confirmations. |
| 24 | Participant communication | Participants do not know which symptoms require urgent contact. | Give clear safety instructions, wallet cards where applicable, and after-hours contact routes. | Participant education documentation. |
| 25 | Vendor oversight | PV vendor processes cases, but sponsor lacks quality visibility. | Track KPIs, reconciliation, case quality, query aging, and compliance metrics. | Vendor oversight report. |
| 26 | Inspection readiness | Safety decisions cannot be reconstructed during audit. | Maintain narratives, source support, correspondence, submission proof, and reconciliation evidence. | Inspection-ready safety file. |
| 27 | Database lock | Unresolved safety discrepancies remain near final analysis. | Close AE, SAE, medication, lab, death, and discontinuation reconciliation before lock. | Final reconciliation sign-off. |
| 28 | Protocol amendments | New safety language is approved but sites keep using old procedures. | Require amendment training, version control, re-consent assessment, and implementation dates. | Amendment training and re-consent tracker. |
| 29 | Benefit-risk review | Safety data are reviewed without connection to efficacy, exposure, or population risk. | Integrate safety trends with endpoint data, dose exposure, and protocol population. | Benefit-risk review summary. |
| 30 | Safety culture | Staff fear reporting too much and under-document borderline events. | Teach that over-escalation can be corrected, while hidden safety data can harm participants. | Rising quality of AE documentation and fewer late SAEs. |
2. Building a Pharmacovigilance Workflow That Works Before the First SAE
The safest trials build pharmacovigilance before the first participant is screened. The sponsor, CRO, PV vendor, medical monitor, CRA team, data management group, and sites should agree on who receives safety information, who assesses it, who enters it, who reconciles it, who submits it, and who follows it until closure. Ambiguity at startup becomes delay during crisis. A strong workflow connects the protocol, safety management plan, monitoring plan, data management plan, medical monitoring plan, and vendor oversight plan. This is the operational backbone behind clinical trial sponsor responsibilities, clinical research project planning, vendor management in clinical trials, and clinical trial resource allocation.
A practical PV workflow should begin with intake. The site must know what information is minimally required for an initial SAE report: identifiable patient, identifiable reporter, suspect product, and adverse event, plus available dates, seriousness criteria, outcome, relevant medical history, concomitant medication, and investigator assessment. The PV team can collect follow-up later, but the initial report should move quickly. CRAs should reinforce this during investigator site management, CRA GCP compliance training, clinical trial protocol management, and informed consent procedures.
The next step is case processing quality. A safety case should be coded consistently, medically reviewed when required, queried intelligently, followed until the outcome is known, and reconciled against clinical databases. Poor case processing creates downstream damage: inconsistent MedDRA coding, missing narratives, unresolved follow-up, weak causality, duplicate cases, late submissions, and unreliable aggregate review. PV teams should treat each case as both a patient story and a data point. That mindset strengthens pharmacovigilance case processing, signal detection, aggregate safety reporting, and regulatory submissions in pharmacovigilance.
Reconciliation should be scheduled, not improvised. The safety database, EDC, lab database, medical coding outputs, death records, discontinuation logs, and concomitant medication data should be compared routinely. A hospitalization in EDC should match the safety database. A medication started for nausea should prompt review of the AE log. A death should match SAE records, narratives, endpoint adjudication, and regulatory reporting. Reconciliation protects clinical data management, EDC systems, clinical data management systems, and clinical trial documentation quality.
A strong workflow also defines escalation routes. Some events require medical monitor review, some require sponsor safety physician input, some require expedited regulatory reporting, some require DMC review, and some require protocol or consent updates. Sites should never have to guess who to contact after hours, during weekends, or when the PI is unavailable. The escalation map should name primary contacts, backup contacts, response time expectations, reporting portals, email templates, phone numbers, and documentation locations. This supports medical monitor oversight, DMC roles, regulatory authority expectations, and clinical research ethics resources.
3. Site-Level Safety Monitoring: Where Most Pharmacovigilance Quality Is Won or Lost
Site-level safety monitoring succeeds when the team treats every visit as a structured safety review. The CRC should ask targeted symptom questions, verify hospitalizations and urgent care visits, review medication changes, check labs, inspect diaries or ePRO entries, confirm dose changes, and document participant complaints in language that can be medically interpreted. The PI should review safety data promptly and document judgment rather than giving verbal approval that leaves no audit trail. This practical discipline sits at the center of CRC responsibilities, patient safety oversight, AE handling for PIs, and GCP compliance for coordinators.
The site should document safety events with enough precision to support causality and follow-up. Poor notes such as “patient felt bad” or “went to hospital” force avoidable queries. Strong source captures onset date, stop date, diagnosis if known, symptoms, severity, seriousness, relationship to study drug or procedure, action taken with study treatment, treatment given, outcome, and whether the event caused withdrawal or dose adjustment. This source discipline supports clinical trial documentation, CRF completion best practices, CRA documentation techniques, and research assistant data collection.
Sites also need strong safety calendars. Required labs, ECGs, pregnancy tests, physical exams, imaging, vital signs, symptom questionnaires, and follow-up calls must happen inside protocol windows. Safety risk increases when assessments happen late, staff miss pre-dose requirements, or sites treat follow-up calls as optional. The CRA should monitor visit windows, recurring missed safety assessments, late PI review, and delayed EDC entry as early warning signs. This connects safety monitoring to protocol management, protocol deviations, clinical trial monitoring terms, and CRA monitoring skills.
Participant education is another safety control. Participants should know which symptoms require immediate reporting, which medications require approval, what to do after hospitalization, how to reach the site after hours, and how to report pregnancy or emergency care. Consent forms may contain safety language, but participants often need simpler instructions they can use when something happens at home. Strong participant education supports informed consent essentials, clinical trial patient education resources, clinical trial ethics, and GCP informed consent procedures.
CRA oversight should focus on safety process health, not field completion alone. During monitoring, the CRA should compare source, AE logs, SAE forms, EDC, medication records, labs, hospital records, and correspondence. The CRA should verify whether PI review was timely, whether causality was documented, whether follow-up is complete, whether safety queries are aging, and whether repeated deviations show training failure. This level of review strengthens clinical trial auditing, inspection readiness, GCP self-assessment, and CRA career skills.
Where is your clinical trial safety monitoring weakest right now?
Choose the safety gap that would create the most risk if an SAE occurred today.
4. Signal Detection, Medical Review, and Aggregate Safety Oversight
Signal detection begins when individual cases are viewed as part of a larger safety pattern. A single headache may mean little. Recurrent severe headaches in a defined population, clustered around dosing, accompanied by hypertension or neurologic findings, can become a signal that deserves investigation. PV teams should evaluate seriousness, frequency, severity, timing, exposure relationship, biological plausibility, risk factors, comparator patterns, and consistency across sites. This is why signal detection and management, biostatistics in clinical trials, primary and secondary endpoints, and DMC safety oversight should work together.
Medical review should challenge safety data intelligently. The medical monitor or safety physician should ask whether event terms are clinically accurate, whether narratives explain the case, whether causality is supported, whether action taken with study product makes sense, whether rechallenge or dechallenge information exists, and whether follow-up is sufficient. A case narrative should tell the medically relevant story without drowning reviewers in unrelated chart noise. That level of review strengthens medical monitor role mastery, medical monitor adverse event reviews, clinical trial medical oversight, and pharmacovigilance specialist terms.
Aggregate review should answer deeper questions than how many cases occurred. The team should ask whether the safety profile is changing, whether risk differs by dose, duration, age, sex, comorbidity, geography, concomitant medication, site behavior, or trial phase. The team should consider whether investigator training, protocol amendments, consent updates, additional monitoring, risk minimization steps, or DMC review are needed. Aggregate reporting becomes valuable when it supports decisions, not when it repeats listings. This connects aggregate reports in pharmacovigilance, risk management plans, clinical trial risk management, and regulatory submissions.
DMC and safety committee packages should be built for judgment. Dumping raw listings on reviewers forces them to hunt for meaning. Better packages show exposure, demographics, withdrawals, deaths, SAEs, AESIs, lab trends, dose modifications, protocol deviations, unblinding concerns, and emerging safety patterns in a structured way. The CRA, medical monitor, statistician, and PV lead should help ensure site-level documentation can support committee-level decisions. This is where DMC roles, randomization techniques, blinding in clinical trials, and placebo-controlled trial interpretation matter.
Safety monitoring should also trigger controlled change. If a signal is credible, the study may need protocol clarification, consent revision, additional labs, modified stopping rules, enhanced site training, investigator letters, IRB notifications, or regulatory submissions. Teams should avoid slow committee drift when participant protection requires action. Strong change control connects clinical trial amendments, informed consent procedures, regulatory submissions in PV, and clinical research regulatory guidelines.
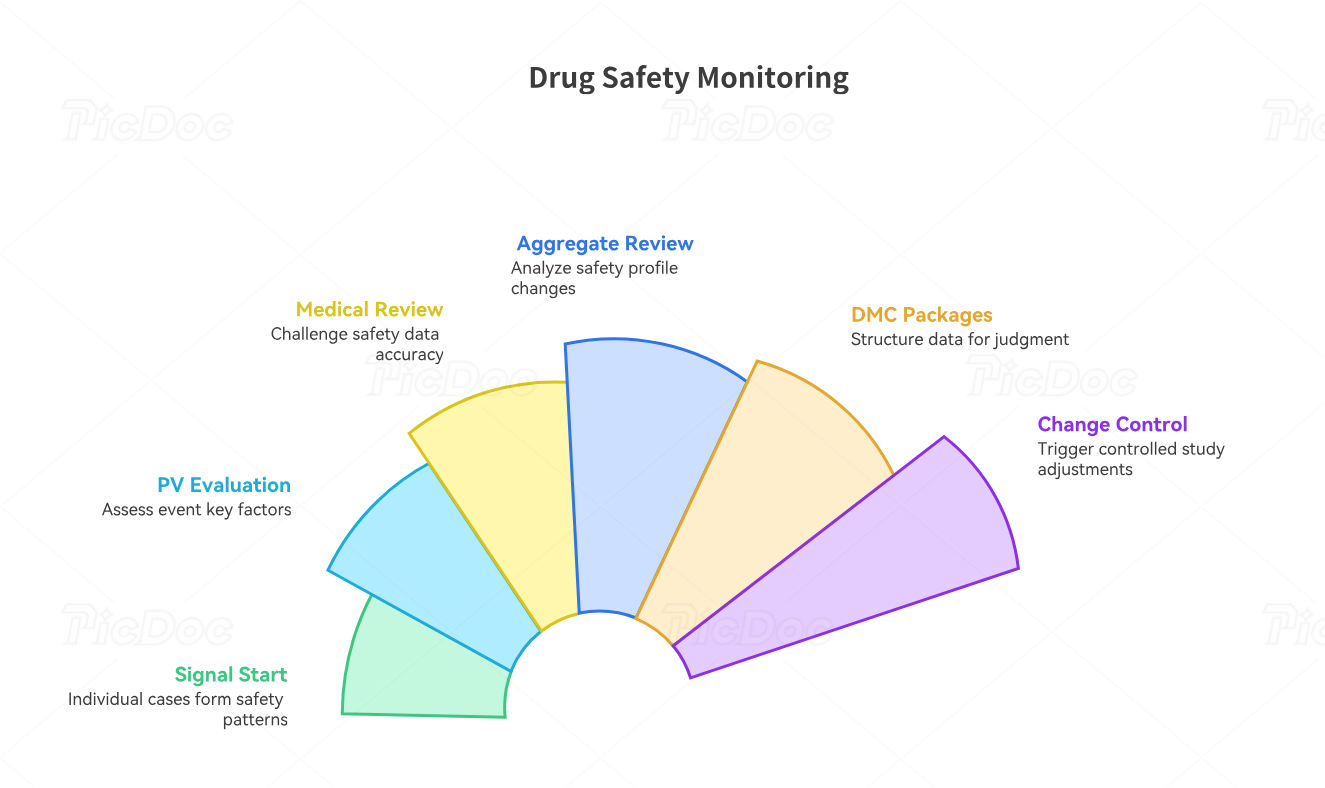
5. Inspection-Ready Pharmacovigilance: Documentation, Reconciliation, and Risk Controls
Inspection-ready pharmacovigilance means every important safety decision can be reconstructed. Inspectors may ask when the site became aware of an SAE, when the sponsor received it, who assessed causality, what follow-up was requested, when the report was submitted, whether the event matched EDC, whether the IB or consent needed updates, and whether similar cases were reviewed. Teams that rely on memory, scattered emails, or undocumented calls create avoidable findings. Strong documentation aligns with clinical trial auditing, clinical trial documentation under GCP, regulatory document management, and ICH guideline expectations.
A safety file should contain safety management procedures, reporting plans, contact lists, training records, SAE forms, narratives, follow-up requests, submission confirmations, reconciliation reports, medical review notes, signal review decisions, DMC correspondence, investigator safety letters, amendment training, and CAPA evidence. The goal is controlled traceability. A reviewer should be able to move from source to EDC to safety database to regulatory submission without losing dates, rationale, or ownership. This supports GCP compliance essentials, handling clinical trial audits, clinical research ethics and compliance, and clinical trial templates and SOPs.
Risk controls should be updated when evidence changes. If one site has repeated late SAE reporting, retrain and monitor effectiveness. If several sites miss AESIs, revise training and source prompts. If lab abnormalities are under-assessed, strengthen PI review procedures. If reconciliation repeatedly finds mismatches, revise database workflows and ownership. CAPA should be targeted, measurable, and followed through. This is where protocol deviation CAPA, GCP deviation management, interactive GCP self-assessment, and clinical trial PM risk management become safety tools.
Vendor oversight is a major PV inspection risk. Sponsors may outsource case processing, medical writing, call center intake, database management, or regulatory submissions, but accountability remains active. Oversight should include case quality review, compliance metrics, reconciliation outcomes, query aging, late case analysis, duplicate detection, narrative quality, submission evidence, and escalation logs. A sponsor that receives polished monthly dashboards but misses unresolved safety defects is still exposed. Strong vendor oversight connects vendor management in clinical trials, pharmacovigilance software review, clinical trial project management, and clinical trial budget oversight.
The best safety programs develop a reporting culture where staff escalate early, document clearly, and ask medical questions quickly. Underreporting often comes from fear of being wrong, fear of creating work, or confusion about definitions. Training should make one principle clear: a possible safety event deserves timely review, and the team can later determine classification, causality, expectedness, and follow-up. That culture protects participants and strengthens clinical research training, clinical research certification, pharmacovigilance career preparation, and clinical research professional development.
6. FAQs About Clinical Trial Safety Monitoring and Pharmacovigilance Best Practices
-
Clinical trial safety monitoring is the structured process of collecting, assessing, documenting, reporting, reconciling, and trending participant safety information during a study. It includes AE capture, SAE reporting, medical review, signal detection, regulatory reporting, DMC review, participant protection, and inspection-ready documentation. It requires coordination between sites, sponsors, CRAs, PIs, medical monitors, PV teams, vendors, and data management. Strong safety monitoring connects adverse event management, SAE reporting, pharmacovigilance principles, and GCP compliance.
-
An adverse event is any unfavorable medical occurrence in a participant, whether or not it is considered related to the investigational product. A serious adverse event meets seriousness criteria such as death, life-threatening risk, hospitalization, disability, congenital anomaly, or other medically important conditions. The operational risk is delay: staff may recognize that something happened but hesitate to escalate because they are unsure whether seriousness applies. Training should reinforce AE identification, SAE procedures, drug safety timelines, and PI adverse event handling.
-
Responsibility is shared, but each role has a specific safety function. The PI provides medical oversight and causality assessment. The CRC captures and documents safety information. The CRA verifies process quality and documentation. The sponsor maintains overall safety accountability. The PV team processes and reports cases. The medical monitor reviews complex events and emerging risks. Data management supports reconciliation. This role clarity depends on PI responsibilities, CRC responsibilities, CRA responsibilities, and medical monitor oversight.
-
The most common mistakes include late SAE reporting, incomplete AE source notes, weak causality rationale, missing follow-up, inconsistent EDC-safety reconciliation, poor lab abnormality review, unclear AESI handling, undocumented PI oversight, weak vendor oversight, and reactive signal detection. These mistakes usually come from workflow gaps rather than a lack of effort. Strong prevention depends on pharmacovigilance case processing, aggregate reporting, signal management, and clinical trial documentation.
-
CRAs can improve safety monitoring by reviewing source notes, AE logs, SAE forms, EDC entries, medication changes, lab abnormalities, hospital records, and PI review timing together. They should look for hidden safety data, late reporting, incomplete follow-up, unresolved medical questions, and repeated safety-related deviations. The strongest CRAs track patterns across visits instead of treating each finding as isolated. This approach strengthens CRA monitoring techniques, CRA documentation skills, inspection readiness, and GCP compliance for CRAs.
-
Signal detection is the process of identifying possible new or changing safety risks from individual cases, grouped patterns, clinical trends, lab shifts, dose relationships, population factors, and cumulative evidence. It requires medical judgment, data review, statistical awareness, and clear documentation of decisions. A signal may lead to enhanced monitoring, protocol changes, consent updates, regulatory communication, or further investigation. Strong signal work connects signal detection mastery, biostatistics in clinical trials, DMC oversight, and risk management plans.
-
The team should maintain clear evidence for AE and SAE collection, reporting timelines, causality assessment, follow-up, reconciliation, medical review, signal review, regulatory submissions, training, DMC decisions, and CAPA. Inspectors should be able to trace a safety event from site awareness through sponsor review, database entry, reporting decision, submission evidence, and follow-up closure. This preparation supports clinical trial audit readiness, CRA inspection readiness, regulatory document management, and ICH guidelines.