
Conducting Investigator Meetings: Comprehensive CRA Strategies
Investigator meetings shape whether a study begins with clean execution or months of preventable rescue work. A CRA who treats the meeting as a compliance checkpoint misses the real opportunity: aligning investigators, coordinators, vendors, safety teams, and sponsor expectations before enrollment pressure exposes every weak handoff. Strong meetings connect GCP compliance, protocol management, site documentation, adverse event handling, and inspection readiness into one operating rhythm.
1. What Investigator Meetings Must Accomplish Before the First Patient Is Enrolled
A high-value investigator meeting should leave every site with the same working picture of the study: who does what, which data points are critical, where the protocol can fail, how safety events move, how deviations are prevented, and what documentation must prove later. This is where a CRA connects the protocol to real site behavior, especially when investigators are busy, coordinators are overextended, and vendors assume the site already understands the technology stack. The CRA should use the meeting to reinforce clinical research associate responsibilities, site qualification expectations, principal investigator oversight, and clinical trial documentation under GCP without turning the session into a passive slide review.
The first objective is protocol translation. Sites need to understand the protocol as workflow, workload, risk, and evidence. A PI may understand the science, while the CRC may struggle with visit windows, source documentation expectations, IP accountability, EDC timing, lab processing, or escalation thresholds. A CRA should make the protocol operational by linking each major procedure to informed consent requirements, case report form quality, primary and secondary endpoints, and protocol deviation prevention.
The second objective is role clarity. Investigator meetings often fail when everyone attends, signs the training log, and leaves with vague confidence. The PI must know what oversight looks like beyond delegation. The sub-investigator must know which medical judgments require documentation. The CRC must know where source notes must be specific. The pharmacist must know how randomization, blinding, storage, temperature excursions, returns, and accountability are controlled. The CRA should repeatedly connect responsibilities to delegation and training expectations, randomization techniques, blinding controls, and GCP inspection preparation.
The third objective is risk rehearsal. A strong CRA does more than explain what should happen. The CRA asks what will happen when a participant arrives outside the visit window, a lab kit is missing, an ePRO entry is incomplete, a potential SAE is identified after clinic hours, a prohibited medication appears in the chart, or a coordinator enters inconsistent endpoint data. These scenarios expose weak points early and connect the investigator meeting to serious adverse event reporting, drug safety timelines, risk management in clinical trials, and clinical trial medical oversight.
| # | Meeting Area | Site Failure Mode | CRA Strategy | Evidence to Capture |
|---|---|---|---|---|
| 1 | Protocol objectives | Site memorizes inclusion rules without understanding endpoint pressure. | Tie each visit activity to endpoint integrity. | Annotated protocol notes and Q&A log. |
| 2 | Eligibility review | Screen failures rise because borderline criteria are handled inconsistently. | Use real eligibility scenarios and force documentation decisions. | Scenario answers and escalation pathway. |
| 3 | Consent workflow | Consent is treated as a signature event instead of a process. | Review timing, version control, comprehension checks, and re-consent triggers using informed consent fundamentals. | Training log plus consent workflow checklist. |
| 4 | PI oversight | PI delegates tasks but fails to document active supervision. | Define what oversight notes, safety review, and protocol decisions must show. | Delegation log, PI meeting cadence, oversight plan. |
| 5 | Delegation log | Staff perform tasks before documented training or authorization. | Cross-check duties against training completion before activation. | Final delegation log and role-specific training proof. |
| 6 | Visit schedule | Window violations occur because coordinators lack calendar controls. | Build visit-window decision trees and backup scheduling rules. | Visit calculator, escalation notes, site calendar controls. |
| 7 | Source documentation | Source lacks rationale, timing, medical judgment, or ALCOA strength. | Show examples of acceptable source notes linked to study documentation skills. | Source template review and site-specific notes. |
| 8 | EDC entry | Data are entered late, inconsistently, or without query discipline. | Set expected entry timelines and query ownership using EDC workflow awareness. | Data entry SLA and query resolution plan. |
| 9 | CRF completion | CRF data conflict with source or endpoint definitions. | Map CRF fields to source requirements and protocol definitions. | CRF-source mapping notes. |
| 10 | Safety reporting | AEs are under-collected because staff wait for patients to volunteer symptoms. | Train active AE probing and documentation using AE identification standards. | AE collection script and medical review pathway. |
| 11 | SAE escalation | Site loses hours deciding whether an event is reportable. | Clarify seriousness, causality, expectedness, and reporting clock triggers. | Emergency contact tree and SAE worksheet. |
| 12 | Protocol deviations | Preventable deviations repeat because root cause is shallow. | Teach deviation triage, CAPA logic, and sponsor notification rules. | Deviation decision tree and CAPA template. |
| 13 | IP accountability | Drug storage, dispensing, returns, or reconciliation create audit risk. | Walk through accountability from receipt to destruction or return. | Pharmacy checklist and temperature log process. |
| 14 | Randomization | Unblinding risk increases through casual access or unclear role separation. | Define randomization permissions, blind protection, and emergency code break rules. | IWRS access list and blind maintenance record. |
| 15 | Labs and specimens | Samples miss processing, shipping, or stability requirements. | Rehearse sample collection timing, kit inventory, centrifuge steps, and courier backup. | Lab workflow sheet and shipping escalation contacts. |
| 16 | Vendor systems | Site staff assume portals work until first patient visit exposes access failures. | Require login verification, role checks, and test transactions before activation. | System access tracker and vendor training proof. |
| 17 | Patient recruitment | Recruitment promises exceed real referral capacity. | Pressure-test recruitment sources using CRC recruitment strategy. | Recruitment plan and screening funnel assumptions. |
| 18 | Retention | Participants drop because burden was underestimated. | Discuss visit length, reminders, reimbursement, transportation, and missed-visit recovery. | Retention plan and participant contact workflow. |
| 19 | Regulatory binder | Documents exist, but filing logic cannot survive inspection. | Align binder sections with regulatory document control. | Binder QC checklist and filing responsibility map. |
| 20 | IRB communication | Amendments, safety reports, and approvals are tracked casually. | Clarify IRB submission triggers, approval dependencies, and version control. | IRB tracker and approval confirmation workflow. |
| 21 | Monitoring expectations | Site treats monitoring as cleanup instead of ongoing quality control. | Set expectations for remote review, SDV readiness, and action item closure. | Monitoring communication plan. |
| 22 | Action items | Meeting issues disappear because owners and due dates are vague. | Close every open question with owner, deadline, and follow-up route. | Action item tracker and minutes. |
| 23 | Training documentation | Attendance exists, but comprehension is weak. | Add knowledge checks and role-based scenarios after core modules. | Attendance log, quiz results, scenario responses. |
| 24 | Hybrid attendance | Remote staff multitask and miss critical operational rules. | Use polling, callouts, breakout discussions, and post-meeting verification. | Attendance analytics and follow-up attestations. |
| 25 | Global sites | Local regulatory or cultural differences create inconsistent execution. | Separate global non-negotiables from country-specific workflows. | Country addendum and local escalation list. |
| 26 | Medical monitoring | Clinical questions bypass the right medical reviewer. | Define when to escalate to sponsor medical monitor using medical monitor responsibilities. | Medical escalation pathway. |
| 27 | Amendments | Sites implement changes before approval or training is complete. | Explain amendment freeze points, effective dates, and re-training requirements. | Amendment implementation tracker. |
| 28 | Audit readiness | Site thinks inspection preparation begins after enrollment. | Build audit-readiness habits from day one using CRA inspection readiness. | Essential document QC plan. |
| 29 | Communication cadence | Sites escalate late because sponsor expectations are unclear. | Set weekly touchpoints, urgent reporting channels, and escalation response times. | Communication plan and contact matrix. |
| 30 | Meeting closeout | Everyone leaves informed, but nobody is operationally ready. | End with readiness gaps, next steps, and activation blockers. | Meeting minutes, tracker, readiness sign-off. |
2. Pre-Meeting Strategy: How CRAs Build a Meeting That Actually Changes Site Behavior
The best CRA work happens before the meeting begins. Preparation should start with the protocol risk profile: complex eligibility, unusual safety reporting expectations, narrow visit windows, central lab fragility, investigational product controls, high-volume data capture, or endpoint assessments that require consistent judgment. A CRA should read the protocol like a future monitor, then build meeting priorities around the errors most likely to create queries, deviations, safety delays, consent findings, and incomplete source. This connects the investigator meeting to clinical trial protocol development, protocol amendments, clinical trial monitoring terms, and GCP guidelines mastery.
The CRA should segment attendees by what they need to perform. PIs need oversight expectations, medical judgment boundaries, safety review duties, and sponsor escalation rules. CRCs need scheduling controls, source templates, EDC requirements, recruitment assumptions, and participant communication workflows. Pharmacists need investigational product storage, accountability, blinding, returns, destruction, and temperature excursion rules. Regulatory staff need document collection, IRB tracking, financial disclosures, version control, and training records. This role-based preparation strengthens CRC responsibilities, research assistant documentation skills, principal investigator responsibilities, and vendor management in clinical trials.
Agenda design should move from study intent to site execution. Weak agendas follow the slide deck order. Strong agendas follow operational consequence: consent before screening, eligibility before randomization, safety before data entry, source before CRF completion, deviation prevention before monitoring, and action items before closeout. A CRA should request time for scenario discussion because scenarios reveal whether the site can apply the protocol under pressure. Examples should include late safety discovery, out-of-window visits, incomplete labs, contradictory medical history, dose hold ambiguity, and PI unavailability. This gives the meeting practical weight through adverse event handling, informed consent best practices, clinical data management, and protocol deviation management.
Pre-meeting outreach also matters. CRAs should contact high-enrolling sites, inexperienced sites, and sites with prior quality concerns before the meeting. Ask what they are worried about, which systems they have used before, whether staff turnover is expected, and whether the PI can attend the full session. A CRA can prevent major activation delays by identifying missing access, expired GCP training, incomplete regulatory documents, weak recruitment assumptions, or unavailable pharmacy support before the first formal session. This belongs inside the same quality mindset as site monitoring visits, site selection visits, clinical trial resource allocation, and clinical trial start-up checklists.
A CRA should enter the meeting with a “must-land” list. These are the five to ten instructions that cannot be buried inside slides: who can consent, when labs must be processed, when SAE clocks start, what creates a major deviation, how randomization is controlled, where source must support endpoint data, how queries must be answered, and how urgent sponsor communication works. The CRA should repeat these points across PI, CRC, pharmacy, and vendor sessions because every site role hears risk differently. This improves sponsor expectations, data monitoring committee awareness, clinical trial project planning, and CRA exam-level monitoring judgment.
3. Running the Meeting: Facilitation Moves That Keep Investigators, CRCs, and Site Teams Engaged
During the meeting, the CRA should listen for false confidence. Sites often nod through protocol procedures because the slides sound familiar, then struggle when one criterion has an exception, one assessment has a timing restriction, or one safety term changes the reporting path. The CRA’s job is to slow down at the points where misunderstanding becomes patient risk, endpoint damage, or inspection exposure. Good facilitation uses targeted questions: “Where will this be documented?”, “Who reviews this before randomization?”, “What happens after hours?”, and “What source proves this decision?” These questions connect training to clinical trial protocol essentials, GCP compliance strategies, source documentation control, and inspection readiness.
The PI session should focus on oversight behavior. A PI who attends the opening scientific presentation and disappears before operational training creates risk. The CRA should reinforce that PI oversight includes reviewing eligibility, assessing medical history, confirming safety causality, documenting clinical judgment, supervising delegated staff, and remaining available for urgent decisions. The strongest CRA approach is specific: show what PI review should look like in source, how safety review should be dated, and when sponsor medical monitor escalation should occur. This strengthens PI clinical trial responsibilities, patient safety oversight, medical monitor AE review, and clinical trial medical oversight.
The CRC session should convert the protocol into visit execution. Coordinators need help seeing where their day can collapse: overlapping visits, missing labs, source fields that cannot be reconstructed, patients who forget diaries, late PI review, open EDC queries, and unclear reimbursement processes. The CRA should walk through the first screening visit, first randomization visit, first safety follow-up, and first monitoring review as if the site is already live. This gives the CRC a real operating model linked to CRC role expectations, patient recruitment mastery, time management for CRCs, and regulatory document management.
The safety section deserves controlled intensity. AEs, SAEs, AESIs, pregnancies, medication changes, hospitalizations, lab abnormalities, and clinically significant findings should be discussed as workflows instead of definitions alone. The CRA should clarify what starts the reporting clock, what information can be submitted before full records are available, who assesses causality, who contacts the sponsor, and how follow-up information is tracked. Sites should leave with zero ambiguity around urgent reporting channels. This is where the meeting protects participants through SAE procedures, drug safety reporting timelines, pharmacovigilance case processing, and pharmacovigilance signal management.
Technology training should focus on friction, not features. EDC, ePRO, IWRS, CTMS, central lab portals, imaging portals, eConsent platforms, and safety portals become study risks when logins, permissions, browser restrictions, user roles, or helpdesk pathways are unresolved. The CRA should confirm which staff need access, which tasks require certification, which systems affect visit flow, and which issues must be escalated before a participant is in the room. This connects investigator meeting execution to EDC systems, CTMS tools, remote patient monitoring tools, and clinical data management systems.
What is the biggest investigator meeting risk on your next study?
Choose the blocker that would create the most downstream cleanup for your CRA or site team.
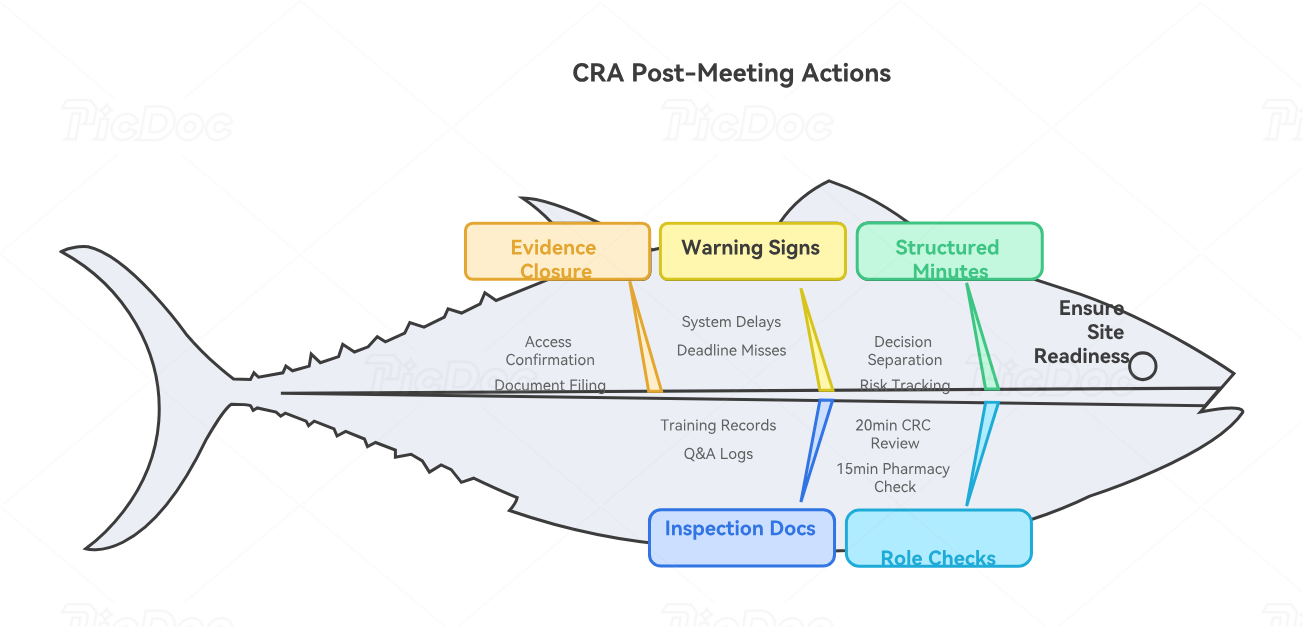
4. Post-Meeting Follow-Up: Turning Attendance Into Site Readiness
The meeting is only successful when the follow-up proves readiness. Attendance logs, slide decks, and certificates show exposure, while action items show whether the site can execute. A CRA should send meeting minutes that separate decisions, open questions, required documents, training gaps, system access issues, activation blockers, and owner-specific deadlines. Every risk discussed during the meeting should reappear in the follow-up tracker. This keeps the study connected to monitoring techniques, documentation control, GCP self-assessment, and audit preparation.
Post-meeting follow-up should verify comprehension by role. The CRA can request completed readiness checklists, site-specific visit flow maps, system access confirmation, pharmacy setup proof, lab kit inventory confirmation, PI oversight plan, and delegation log finalization. For high-risk studies, the CRA should schedule short role-specific touchpoints after the main meeting instead of assuming the general session was enough. A 20-minute CRC workflow review can prevent weeks of query cleanup. A 15-minute pharmacy check can prevent investigational product findings. A PI oversight conversation can prevent documentation gaps that become inspection findings. This reinforces site management mastery, clinical trial resource allocation, risk-based monitoring logic, and clinical research project planning.
A CRA should also look for soft warning signs after the meeting. Sites that delay system access, ask basic questions already covered, miss document deadlines, delegate everything to one overwhelmed coordinator, or cannot confirm PI availability may need intensified support before activation. These signs predict future problems: late data entry, preventable deviations, inconsistent safety reporting, participant retention issues, and monitoring action items that never close. Strong CRAs escalate early with facts, not frustration. They connect each warning sign to sponsor risk, participant risk, or inspection risk using clinical trial sponsor responsibilities, CRC exam practice logic, CRA certification mistakes, and clinical trial budgeting oversight.
Post-meeting documentation must be inspection-minded. Keep the agenda, attendee list, role-based training records, slide deck version, Q&A log, knowledge checks, minutes, follow-up actions, and evidence of completed retraining when needed. If an inspector later asks how the site was trained on a critical amendment, safety procedure, endpoint assessment, or protocol-required device, the study team should be able to reconstruct what was trained, when it was trained, who attended, what questions were answered, and what evidence proves completion. This aligns investigator meeting follow-up with essential training requirements, regulatory document management, clinical research ethics and compliance, and ICH guideline expectations.
The CRA should close follow-up with readiness language that is direct and evidence-based. Instead of saying the site is trained, the CRA should document that required staff completed role-based training, system access was confirmed, investigational product controls were reviewed, safety reporting escalation was verified, essential documents were filed, and open issues have assigned owners. This protects the site and sponsor because activation decisions should rest on proof, not optimism. That is the operational bridge between site start-up activities, clinical trial documentation, GCP compliance for CRAs, and inspection readiness for CRAs.
5. Advanced CRA Strategies for Hybrid, Global, and High-Risk Investigator Meetings
Hybrid investigator meetings require more control because remote attendance creates silent disengagement. A CRA should assume some attendees will multitask unless the meeting forces interaction. Use role callouts, short polls, breakout scenarios, timestamped attendance, post-session knowledge checks, and separate Q&A rooms for CRCs, pharmacists, investigators, and regulatory staff. Remote participants should confirm system access, training completion, and role-specific readiness after the session. This protects execution across virtual clinical trial workflows, remote monitoring tools, ePRO systems, and clinical trial technology innovations.
Global investigator meetings need a layered training model. The CRA should distinguish global protocol requirements from local regulatory processes, ethics committee timelines, source documentation norms, safety submission routes, drug importation issues, language requirements, and country-specific recruitment realities. Sites should understand the non-negotiable study standards and the local pathways that support them. A global meeting becomes dangerous when country differences are treated as footnotes. This is especially important for teams working across clinical research regulatory authorities, global regulatory guidelines, top clinical trial countries, and clinical trial sites in Europe.
High-risk studies require more rehearsal. Oncology trials may need intense AE grading, dose modification, imaging schedules, and rapid escalation pathways. Rare disease trials may require careful eligibility confirmation, caregiver coordination, and retention support. Phase I trials may need dose escalation rules, sentinel dosing controls, and intensive safety oversight. Cardiovascular, neurology, infectious disease, and pediatric studies each bring different endpoint and participant-protection pressures. The CRA should shape investigator meeting content around the therapeutic area’s failure modes using oncology site expertise, neurology trial site capabilities, phase I trial processes, and pediatric clinical trial considerations.
Advanced CRAs also manage meeting politics. Sponsors may want speed, investigators may want scientific discussion, vendors may want system demos, and coordinators may need practical workflows. The CRA often becomes the person who notices that the meeting is polished but operationally thin. The solution is disciplined facilitation: protect time for workflow risks, capture unresolved issues openly, prevent vendor assumptions from becoming site burden, and make sure the PI hears the responsibilities that cannot be delegated away. This connects the CRA’s meeting leadership to stakeholder communication, clinical trial PM strategies, vendor management, and clinical research staffing realities.
The strongest CRA strategy is to treat the investigator meeting as the first quality intervention. Every slide should answer a future monitoring question. Every scenario should prevent a future deviation. Every Q&A response should reduce later confusion. Every action item should move the site closer to clean enrollment. When the meeting works, the first participant visit feels controlled because the site has already practiced the hard parts. That is the standard CRAs should bring to clinical trial monitoring, GCP compliance, protocol deviation control, and clinical research certification preparation.
6. FAQs About Conducting Investigator Meetings as a CRA
-
A CRA should prioritize the operational risks that can damage participant safety, data integrity, protocol compliance, and inspection readiness. The most important areas are eligibility interpretation, informed consent timing, PI oversight, safety reporting, protocol deviation prevention, investigational product control, source documentation, CRF completion, system access, and escalation pathways. A meeting that covers all slides but fails to clarify these areas leaves the site exposed. The CRA should connect every major discussion to GCP compliance, CRA monitoring responsibilities, protocol management, and clinical trial documentation.
-
Experienced investigators respond better to decision points than basic definitions. Use case scenarios involving eligibility conflicts, safety causality, dose holds, prohibited medications, and endpoint assessments. Ask how they would document the decision and when they would escalate. This respects their expertise while exposing gaps that can affect the study. Strong CRA facilitation links investigator engagement to PI responsibilities, patient safety oversight, medical monitor collaboration, and adverse event review.
-
The study team should retain the final agenda, approved slide deck, attendance records, role-based training logs, Q&A log, meeting minutes, action item tracker, knowledge checks, system training proof, and evidence of follow-up training. If an amendment, safety update, or protocol clarification is discussed later, that training should be documented with the same discipline. These records support regulatory document control, GCP training requirements, inspection readiness, and clinical trial audit preparation.
-
Unanswered questions should be documented with the owner, required input, due date, and communication pathway. A CRA should avoid informal answers when the question affects eligibility, safety, dosing, endpoint interpretation, or regulatory obligations. These questions may require sponsor, medical monitor, data management, pharmacovigilance, or regulatory review. The follow-up response should be distributed to affected sites and filed appropriately. This protects sponsor communication, medical oversight, data management quality, and pharmacovigilance reporting.
-
Investigator meetings prevent deviations when they rehearse real site scenarios before enrollment begins. Sites need to know how to manage visit windows, eligibility documentation, consent version control, prohibited medications, missed assessments, lab timing, randomization rules, and safety reporting triggers. A CRA should show where deviations usually begin and how the site can intercept them. This makes the meeting directly useful for protocol deviation corrective actions, GCP deviation management, clinical trial amendments, and CRC protocol responsibilities.
-
A hybrid meeting succeeds when remote attendees are held to the same operational standard as in-person attendees. Require role-specific attendance tracking, interactive questions, system access verification, knowledge checks, documented Q&A, and post-meeting readiness confirmation. Remote staff should prove they can execute the protocol, not merely prove they logged in. This approach supports virtual trial readiness, remote patient monitoring, EDC readiness, and clinical trial technology adoption.
-
A CRA should measure success through readiness evidence: resolved action items, completed training, confirmed system access, finalized delegation logs, clear safety escalation pathways, documented PI oversight expectations, complete regulatory documents, and site confidence during scenario review. Clean first-patient execution is the real test. When early visits produce fewer preventable deviations, fewer avoidable queries, faster safety reporting, and cleaner source, the meeting did its job. This measurement mindset connects to clinical monitoring performance, site documentation quality, clinical trial project planning, and GCP compliance assessment.