
Handling Pharmacovigilance Audits & Inspections: Expert Techniques
A pharmacovigilance audit tests the real nervous system behind patient safety: intake, triage, seriousness assessment, expectedness, timelines, reconciliation, signal escalation, vendor control, training, CAPA, and inspection storytelling. Weak teams prepare documents; strong teams prepare proof chains. The difference matters because PV inspections can examine routine compliance, for-cause concerns, safety reporting failures, and whether the organization can explain every safety decision with evidence. EMA GVP sets the EU pharmacovigilance practice framework, GVP Module IV supports risk-based PV audits, GVP Module III describes risk-based and for-cause PV inspections, FDA BIMO includes inspections, data audits, and remote regulatory assessments, and ICH E2E supports planned pharmacovigilance activity around safety risks.
1. Build Inspection Readiness Around Evidence Trails, Not Document Storage
The strongest pharmacovigilance audit program starts with one question: can an inspector follow a safety event from first awareness to final regulatory action without hitting confusion, missing timestamps, unclear ownership, or undocumented judgment? A team that understands what pharmacovigilance requires, drug safety reporting timelines, clinical trial safety monitoring, and global regulatory compliance in pharmacovigilance prepares by proving control, rather than assembling a last-minute folder that only looks clean from a distance.
Audit readiness has three layers. The first is process accuracy: every SOP must match the way work actually happens across intake, duplicate checks, case validation, medical review, seriousness assessment, causality, submission, follow-up, literature review, reconciliation, and archiving. The second is evidence integrity: timestamps, version histories, email handoffs, vendor confirmations, quality checks, training records, and escalation logs must show that the safety system operated within defined rules. The third is response discipline: when an auditor asks why a decision was made, the team must answer using source data, procedure logic, documented rationale, and risk impact. That same thinking supports aggregate report preparation, regulatory submission mastery, adverse event handling, and medical monitor adverse event review.
The painful audit failures usually begin months before the inspection notice arrives. A site forwards safety information late. A vendor reconciliation gap stays open. A case processor makes an expectedness decision with thin documentation. A medical review comment says “acceptable” without explaining why. A submission confirmation sits in a mailbox instead of the validated system. Each issue feels small until an inspector threads them together and sees a weak safety culture. High-performing PV teams turn those weak points into measurable controls: aging dashboards, monthly reconciliation, CAPA trend reviews, documented quality sampling, role-based training, and escalation thresholds tied to patient risk. That is the same operational mindset behind strong GCP compliance strategies, CRA inspection readiness, clinical trial documentation control, and research compliance mastery.
| Audit Area | What Inspectors Test | Common Failure Mode | Expert Technique |
|---|---|---|---|
| Safety intake | First awareness capture, date stamps, source traceability | Safety information arrives through informal channels | Map intake to pharmacovigilance fundamentals and test every source monthly |
| Case validity | Identifiable patient, reporter, suspect product, event | Invalid cases treated inconsistently | Use validation checklists aligned with drug safety reporting requirements |
| Seriousness assessment | Medical judgment and documented criteria | Hospitalization, disability, and medically important events under-explained | Require reviewer rationale connected to SAE reporting procedures |
| Expectedness | Current reference safety information and version control | Team applies outdated RSI or product information | Maintain controlled references through regulatory document management |
| Causality | Medical rationale, temporal relationship, alternative explanations | “Related” or “unrelated” chosen without reasoning | Use structured review notes from medical monitor AE review |
| Expedited reporting | Clock start, submission deadline, confirmation evidence | Clock start differs across teams | Audit clock logic against safety reporting timelines |
| Follow-up attempts | Medical completeness and persistent outreach | Missing follow-up documentation | Use event-specific templates tied to AE reporting techniques |
| Duplicate detection | Cross-source matching and case linkage | Duplicate cases inflate signals or hide late reports | Run duplicate logic before aggregate report preparation |
| Reconciliation | Safety database versus clinical, medical information, vendor, and site logs | Open discrepancies lack owners | Escalate aging gaps through vendor management controls |
| Literature monitoring | Search strategy, review frequency, relevance decisions | Screening rationale too thin | Document inclusion logic like clinical research publication review |
| Signal detection | Trend review, disproportionality triggers, medical escalation | Signal meetings become passive summaries | Connect trend review to clinical trial safety monitoring |
| Aggregate reports | Data lock, case line listings, cumulative analysis | Case narratives and tables disagree | Use pre-lock QC from aggregate reporting workflows |
| Regulatory submissions | Routing, submission proof, authority-specific expectations | Submission evidence scattered across emails | Centralize receipts using regulatory submission controls |
| Global compliance | Local rules, affiliates, distributors, partner agreements | Local reporting paths undocumented | Create country controls from global PV compliance planning |
| SOP governance | Current procedures, effective dates, implementation proof | SOP updates released without training impact review | Link training completion to GCP training requirements |
| Training | Role-based curricula, overdue training, competency checks | People trained on generic PV slides only | Build role tracks for PV associates and safety reviewers |
| CAPA | Root cause, correction, prevention, effectiveness checks | CAPA closes after correction, before prevention is proven | Test recurrence risk using corrective action discipline |
| Quality metrics | Late cases, correction rates, reopen rates, query aging | Metrics reported without thresholds | Use action triggers like risk-based monitoring |
| Vendor oversight | Contracts, KPIs, QC, escalation, audit rights | Sponsor assumes outsourced means transferred responsibility | Apply sponsor oversight from sponsor responsibility guidance |
| Safety database validation | Access control, audit trails, change control, data integrity | System changes lack business verification | Document data decisions like clinical data review |
| Access management | Least privilege, terminated users, reviewer permissions | Former vendors retain access | Include access checks in clinical research technology governance |
| Business continuity | Backup processes, downtime procedures, emergency reporting | Contingency plans exist but remain untested | Simulate outages before GCP audit preparation |
| Partner agreements | Safety data exchange agreements, timelines, contact lists | Agreement language conflicts with operational practice | Review obligations through stakeholder communication strategy |
| Inspection room control | Request intake, response tracking, document release | Teams answer too quickly with unverified documents | Use controlled response workflows from inspection readiness practices |
| Interview preparation | Role clarity, factual answers, escalation discipline | Staff speculate, over-answer, or contradict SOPs | Train interviewers with scientific communication habits |
| Management oversight | Governance minutes, risk acceptance, resourcing decisions | Leadership sees PV metrics after problems mature | Use dashboards tied to resource allocation and risk ownership |
| Post-inspection response | Observation analysis, CAPA depth, effectiveness proof | Responses promise fixes without evidence plans | Build response packs using clinical quality auditor thinking |
2. Create a Pharmacovigilance Audit File That Tells the Whole Safety Story
A strong PV audit file should read like a controlled safety narrative, starting with governance and ending with proof that patient risk is being monitored, escalated, and reduced. The file should include the PV system overview, process maps, SOP index, training matrix, case processing metrics, late-case logs, reconciliation evidence, vendor oversight records, safety database controls, signal management records, aggregate report schedules, regulatory submission confirmations, CAPA register, deviation trends, and management review minutes. This connects directly to pharmacovigilance career fundamentals, drug safety specialist advancement, PV manager skill development, and regulatory affairs specialist responsibilities.
The highest-value technique is the “traceable sample pack.” Select a small set of cases across seriousness levels, reporting pathways, products, studies, partners, and countries. For each sample, prepare the complete trail: intake record, source document, validation screen, seriousness rationale, causality assessment, expectedness check, follow-up attempts, quality review, submission evidence, reconciliation status, medical review, and any CAPA linkage. This prevents the inspection room from becoming a scavenger hunt. It also exposes gaps early, especially when clinical trial protocol management, site monitoring visit documentation, clinical trial data verification, and remote monitoring workflows feed safety oversight.
Weak audit files often fail through contradiction. The SOP says quality review occurs before submission, while the case record shows review after submission. The vendor agreement says monthly reconciliation, while evidence shows quarterly reconciliation. The training matrix says the reviewer is qualified, while the role description requires medical review competency that was never assessed. The CAPA says recurrence is prevented, while the same late-case pattern appears in the next quarter. Expert teams run contradiction checks before the auditor does. They compare procedure against evidence, metric against source, agreement against performance, and CAPA promise against trend. That discipline strengthens GCP compliance essentials for CRAs, regulatory and ethical PI responsibilities, patient safety oversight, and clinical compliance officer career skills.
3. Control the Inspection Room With Request Discipline, Interview Discipline, and Version Discipline
During an inspection, the best teams slow the room down without appearing defensive. Every request should be logged with the exact wording, requester, due time, owner, source system, version, quality reviewer, release approval, and final delivery status. Document release must be controlled because a wrong draft, outdated SOP, unreviewed spreadsheet, or incomplete case file can create a finding that never existed in the real process. This is why PV inspection readiness should borrow habits from clinical trial audit preparation, CRA audit readiness, documentation management, and clinical quality auditor advancement.
Interview discipline is equally important. Staff should answer the question asked, explain their actual role, describe the governing SOP, show where evidence lives, and escalate when the question crosses into another function. The most damaging interviews usually come from helpful people who speculate beyond their responsibility. A case processor should describe intake and processing controls. A medical reviewer should explain seriousness, causality, expectedness, and medical judgment. A quality lead should explain sampling, deviation review, CAPA, and trend oversight. A vendor manager should explain service-level governance and escalation. That role clarity supports medical monitor AE review, MSL scientific communication, effective stakeholder communication, and clinical operations leadership.
Version discipline protects credibility. Inspectors may ask for current SOPs, prior SOPs, effective-date histories, training assignments, reference safety information, line listings, submission receipts, aggregate reports, and CAPA records. The response team should confirm whether the request relates to the current process, the process active at the time of the event, or a historical trend period. A late SAE from last year should be judged against the SOP and agreement active at that time, while a current system control should be judged against the live procedure. That single distinction can prevent false inconsistency. The same maturity appears in IND application planning, clinical trial amendments, protocol deviation CAPA, and regulatory submission workflows.
What is the biggest PV inspection risk in your current safety system?
Choose one. Your answer points to the control that should be stress-tested first.
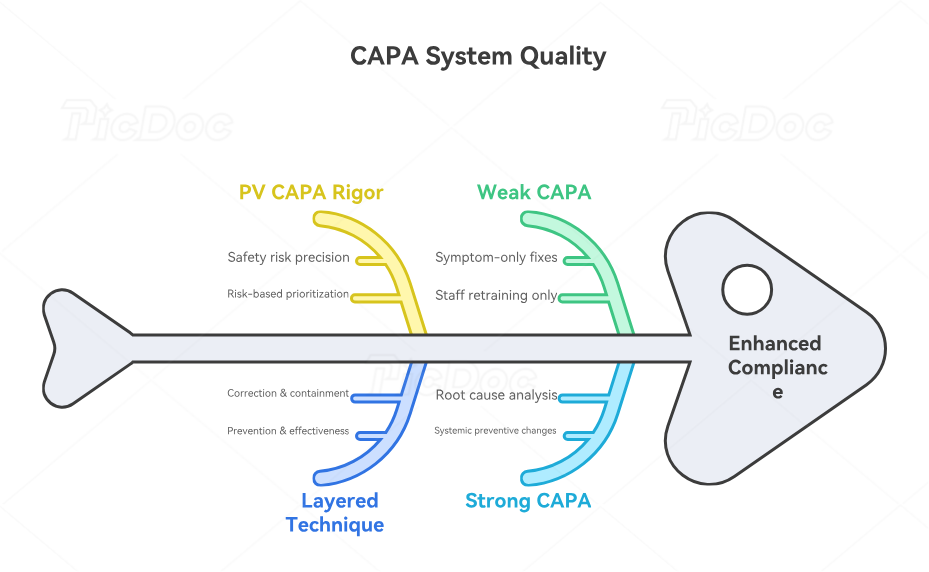
4. Turn CAPA Into Inspection Defense, Not Administrative Cleanup
CAPA is where inspectors learn whether the organization fixes systems or only fixes symptoms. A weak CAPA says, “staff retrained.” A strong CAPA explains the root cause, affected process, risk impact, immediate correction, preventive change, owner, due date, effectiveness measure, evidence source, and recurrence trigger. For pharmacovigilance, CAPA must address safety risk with precision: missed intake pathway, unclear clock-start rule, underpowered vendor oversight, outdated work instruction, poor medical review template, incomplete reconciliation, or metric thresholds that never forced escalation. This mirrors the rigor required in protocol deviation corrective actions, GCP compliance self-assessment, clinical trial audits, and research compliance ethics.
The best CAPA technique is to separate correction, containment, prevention, and effectiveness. Correction handles the specific defect: submit the report, complete follow-up, reconcile the case, correct the narrative, archive the receipt. Containment checks whether the same defect affects other products, studies, vendors, countries, or time periods. Prevention changes the system: revised workflow, automated alert, new QC step, escalation threshold, agreement update, training with competency assessment. Effectiveness proves the issue stayed controlled after implementation. Those layers create inspection-ready evidence and strengthen clinical trial safety monitoring, aggregate report quality, global compliance oversight, and sponsor responsibility controls.
PV CAPA also needs risk-based prioritization. A typo in a non-submitted internal note and a late serious unexpected case require very different urgency. A missed partner reconciliation covering one low-volume product differs from a repeated affiliate reporting failure across multiple countries. Use a scoring model that considers patient risk, regulatory impact, recurrence, product maturity, volume, detectability, and process dependency. Then match CAPA depth to risk. That approach gives leadership a defensible reason for resource allocation and prevents inspection responses from sounding arbitrary. It also aligns with risk-based monitoring strategies, clinical trial resource allocation, vendor management in clinical trials, and clinical project manager governance.
5. Prepare People, Vendors, and Leadership Before the Inspection Notice Arrives
Inspection readiness becomes fragile when only the PV quality team understands it. Case processors, medical reviewers, safety scientists, regulatory affairs, clinical operations, data management, medical information, affiliates, vendors, and leadership all touch the safety system. Each group needs a narrow readiness brief: what they own, what evidence proves it, what metrics reveal weakness, what questions they may receive, and when to escalate. Role-specific readiness works better than generic annual training because inspections test actual work. This connects to essential GCP training, clinical data management career skills, clinical regulatory specialist duties, and quality assurance career development.
Vendors require special attention because outsourced PV work often becomes the most exposed inspection area. The sponsor should be able to show the vendor selection rationale, contract scope, safety data exchange agreement, KPIs, quality sampling, issue escalation, meeting minutes, audit outcomes, CAPA tracking, business continuity testing, and access control. The strongest vendor oversight files show active supervision rather than passive receipt of monthly reports. They also show that performance concerns triggered decisions. When late cases rose, the sponsor increased sampling. When reconciliation gaps aged, leadership escalated. When a quality trend repeated, CAPA moved beyond retraining. Those habits reinforce vendor management essentials, stakeholder communication strategy, clinical operations management, and clinical trial project management.
Leadership preparation matters because inspectors often look for governance, escalation, and resourcing evidence. A VP, safety head, medical lead, or compliance officer should explain how PV risks are surfaced, how late or serious issues reach management, how resources are assigned, how vendors are challenged, and how CAPA effectiveness is reviewed. Leadership should never sound detached from the safety system. Management review minutes, risk registers, trend dashboards, and action logs should show that leadership sees weak signals early. This creates a stronger inspection posture across patient safety oversight, clinical quality auditing, clinical compliance leadership, and regulatory affairs leadership.
6. FAQs: Handling Pharmacovigilance Audits & Inspections
-
Start with the processes that can create patient risk or regulatory exposure: safety intake, case validation, seriousness assessment, expectedness, causality, expedited reporting, follow-up, reconciliation, signal detection, aggregate reporting, and CAPA. Then build traceability around those processes. The goal is to show who did the work, when it happened, what procedure governed it, what evidence proves it, and how exceptions were handled. A practical readiness plan should connect pharmacovigilance basics, drug safety reporting, clinical safety monitoring, and global PV compliance.
-
The most important documents are the ones that prove control over safety-critical decisions. These include SOPs, work instructions, training records, safety database records, case files, submission confirmations, reconciliation logs, signal meeting minutes, aggregate report files, vendor agreements, quality metrics, deviation logs, CAPA records, audit reports, and management review evidence. The inspection team should be able to connect these documents into one coherent story. Strong preparation overlaps with regulatory document management, CRA documentation techniques, regulatory submissions, and aggregate report preparation.
-
Answer with role clarity, evidence, and procedure logic. The person answering should explain only the process they own, reference the governing SOP, show where the evidence sits, and escalate questions that belong to another function. Difficult questions become dangerous when staff speculate or try to fill gaps from memory. A better response sounds like: “My role is case quality review. The medical assessment is completed by the safety physician. I can show the QC checklist, query history, and escalation record.” That discipline is reinforced by scientific communication skills, medical monitor review, stakeholder communication, and inspection preparation.
-
A strong CAPA proves root cause, risk impact, correction, containment, prevention, and effectiveness. It should explain the real system weakness, show whether the issue affected other cases or products, document immediate correction, implement preventive controls, and prove the issue stayed controlled after the fix. “Training completed” is weak when the root cause involves unclear workflow, poor system configuration, vendor underperformance, or missing escalation thresholds. Effective CAPA thinking should connect protocol deviation corrective actions, GCP self-assessment, clinical quality auditing, and PV manager development.
-
A sponsor proves vendor oversight by showing active governance, measurable performance review, issue escalation, and documented decisions. The file should include the safety data exchange agreement, scope of work, KPIs, quality sampling, reconciliation evidence, meeting minutes, audit results, CAPA tracking, escalation logs, and evidence that leadership responded when trends worsened. Vendor oversight should show control before a crisis, not after an observation. This is where vendor management skills, sponsor role clarity, resource allocation, and clinical project management become essential.
-
A PV team should conduct mock inspections often enough to test real risk areas before regulators, partners, or internal auditors find them. High-risk products, new vendors, recent safety system changes, repeated late cases, new markets, major SOP revisions, database migrations, and significant CAPA trends all justify targeted mock inspection activity. The best mock inspections use live samples rather than polished binders. They trace cases, challenge timestamps, interview role owners, test document retrieval, and evaluate whether leadership understands risk. This strengthens risk-based monitoring, remote and on-site monitoring, clinical trial audit readiness, and GCP preparation.