
Understanding IND/NDA Submissions: Regulatory Affairs Mastery
Regulatory affairs mastery begins when a professional stops treating submissions as paperwork and starts treating them as evidence architecture. An IND protects the path into human research; an NDA defends the path to market approval. Every protocol, case report form, clinical trial amendment, serious adverse event, statistical endpoint, and regulatory document either strengthens that story or creates review friction that costs months.
1. Understand the Strategic Difference Between IND and NDA Submissions
An Investigational New Drug application is the regulatory bridge that allows a sponsor to test an investigational drug in humans under FDA oversight. In the U.S., IND requirements sit under 21 CFR Part 312, which governs investigational new drugs, IND submission, FDA review, sponsor responsibilities, protocol amendments, information amendments, and IND safety reporting. A sponsor subject to IND requirements must have the IND in effect before beginning the clinical investigation.
That single point changes how regulatory affairs professionals should think. The IND is built around permission to proceed safely. The NDA is built around proof that the product deserves approval. The IND asks whether the proposed human testing plan is reasonably safe and scientifically justified; the NDA asks whether the accumulated nonclinical, clinical, CMC, statistical, safety, labeling, and risk-benefit evidence supports marketing approval. That is why GCP compliance, clinical trial protocol management, clinical trial sponsor responsibilities, and patient safety oversight must be designed with the eventual NDA in mind.
FDA describes the NDA as documentation that tells the drug’s “whole story,” including clinical testing, ingredients, animal study results, behavior in the body, and manufacturing, processing, and packaging information. That “whole story” language is important because weak submissions often fail through fragmentation. Clinical teams may know the study history, safety teams may know the adverse event profile, CMC teams may know manufacturing evolution, and statisticians may know endpoint logic, yet the submission collapses if those pieces appear disconnected.
Regulatory affairs professionals therefore sit at the intersection of science, operations, formatting, and persuasion. They need enough biostatistics literacy to understand endpoint defensibility, enough pharmacovigilance understanding to recognize safety narrative risk, enough CRA monitoring awareness to understand site data reliability, and enough regulatory career discipline to keep the submission from becoming a last-minute rescue operation.
2. Build IND Readiness Before the Application Becomes an Emergency
A strong IND submission starts before the first official publishing timeline appears. The regulatory affairs team should begin by asking three questions: why this product, why this population, and why this protocol now? Those answers need to appear consistently across the investigator brochure, protocol, informed consent, CMC information, nonclinical package, and safety monitoring plan. When teams build the IND this way, investigational new drug application preparation becomes a controlled process instead of a scramble around missing narratives.
FDA’s IND forms page lists core forms used in IND work, including Form FDA 1571 for the IND application, Form FDA 1572 for investigator statements, Form FDA 3674 for clinical trial certification compliance, and financial disclosure forms. Those forms look administrative, but they represent accountability. The person named as sponsor carries responsibility. The investigator statement connects human research conduct to named investigators. The financial disclosure package protects credibility around clinical investigator bias. A regulatory professional who treats forms as “clerical” creates risk for the entire clinical program.
The IND package should also show that the proposed trial can run cleanly. That requires alignment between the clinical research coordinator role, site monitoring visit expectations, research compliance ethics, and GCP training requirements. A trial can have an elegant scientific premise and still create regulatory pain if visit windows, safety assessments, lab handling, source documentation, or eligibility confirmation are too loose for real sites to execute.
Safety planning deserves special attention. FDA’s IND safety reporting resource states that sponsors must report serious and unexpected suspected adverse reactions, and unexpected serious suspected adverse reactions must be reported as soon as possible, within 15 calendar days after initial sponsor receipt. That timeline should shape the sponsor’s safety intake workflow, site training, medical review routing, narrative authoring, and submission calendar. It also affects how teams train investigators on adverse event handling, drug safety reporting, medical monitor review, and pharmacovigilance career skills.
The IND also needs realistic amendment discipline. Every major protocol change should be evaluated for impact on participant safety, endpoint validity, operational feasibility, consent language, site training, monitoring, database design, and statistical interpretation. The painful truth is simple: many NDA problems begin as IND-stage decisions that nobody tracked carefully. A rushed protocol change can later create explainability gaps in protocol deviations, clinical trial data verification, handling clinical trial audits, and risk-based monitoring.
3. Prepare the NDA Like a Reviewer Will Test Every Claim
The NDA is where evidence becomes an approval argument. Under 21 CFR Part 314, the drug approval process is designed to facilitate approval of drugs shown to be safe and effective and ensure disapproval of drugs that have not been shown to be safe and effective. That means regulatory writing must connect every claim to a specific body of evidence. If the proposed indication says one thing, the pivotal population says another, and the efficacy tables imply something narrower, reviewers will feel the gap immediately.
NDA preparation should begin during late-stage development, long before final tables are ready. Regulatory affairs should create a claim-evidence matrix that links the proposed indication, dosing regimen, primary endpoints, secondary endpoints, safety profile, subgroup interpretation, CMC readiness, and labeling language. This is where primary and secondary endpoints, placebo-controlled trial design, randomization methods, and blinding strategy become more than trial design topics; they become the backbone of the approval story.
The most dangerous NDA mistake is overclaiming from incomplete alignment. A product may show a meaningful treatment effect, but the submission still needs to explain missing data, protocol deviations, analysis populations, exposure, adverse events of special interest, dose modifications, discontinuations, and population limitations. That is why a regulatory team should work closely with clinical data managers, clinical data coordinators, lead clinical data analysts, and clinical quality auditors before integrated summaries become fixed.
The CMC section also needs executive-level attention. Many clinical teams underestimate how severely manufacturing, controls, specifications, stability, comparability, and process changes can affect review confidence. A submission can have strong clinical outcomes and still face delay if the product quality story feels immature. Regulatory affairs must keep CMC, clinical, safety, and labeling connected so the proposed commercial product matches the product studied. That connection matters for regulatory affairs associates, clinical regulatory specialists, clinical compliance officers, and quality assurance specialists.
NDA labeling should be treated as a controlled negotiation with evidence. The indication, dosage and administration, warnings, precautions, adverse reactions, use in specific populations, and clinical studies sections all need source-backed discipline. Labeling ambition without data support creates review conflict. Conservative labeling without strategic clarity can weaken commercial value. The best regulatory professionals understand both sides and prepare the team with effective stakeholder communication, clinical project management, vendor management, and regulatory submission mastery.
What is your biggest IND/NDA submission blocker right now?
Choose one. Your answer points to the fastest regulatory affairs fix.
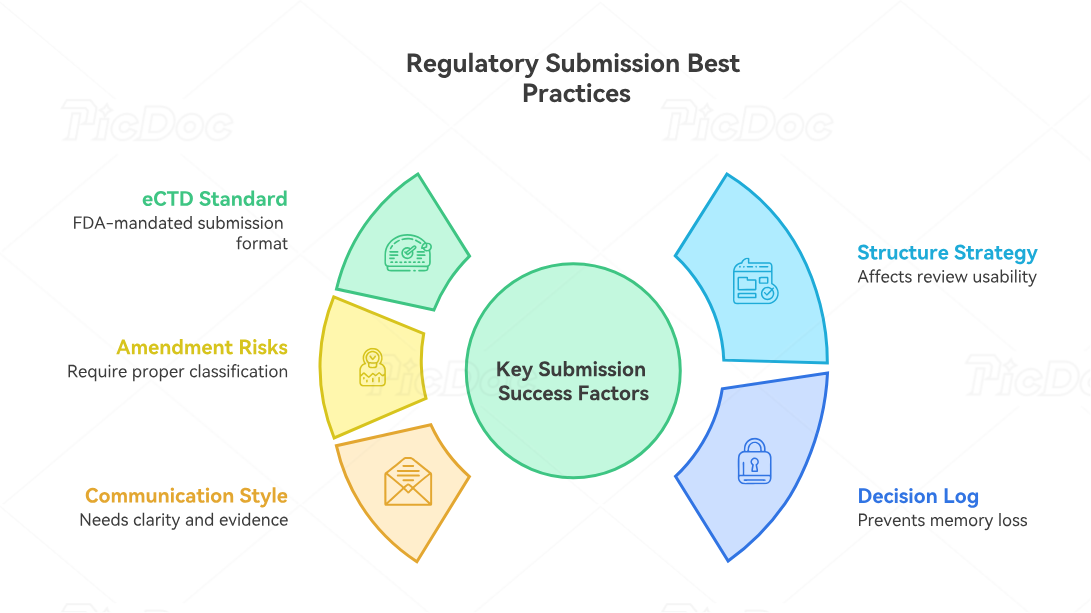
4. Master the eCTD, Amendment, and Communication Workflow
Submission quality depends on the content and the container. FDA identifies eCTD as the standard format for submitting applications, amendments, supplements, and reports to CDER and CBER. This matters because a strong scientific package can lose reviewer confidence when navigation is messy, lifecycle management is poor, cross-references are broken, or old files remain active where replaced documents should control the record.
Regulatory affairs teams should treat eCTD structure as part of submission strategy. Module placement, document granularity, naming conventions, hyperlinks, bookmarks, leaf lifecycle, version control, and sequence planning all affect review usability. The goal is to help reviewers find the right evidence at the right moment. That requires disciplined collaboration among regulatory operations, medical writing, clinical operations, safety, statistics, CMC, and quality. Professionals who understand directory of clinical trial templates, interactive GCP compliance checks, trial startup checklists, and clinical research technology adoption are better prepared for this operational reality.
Amendments create a second layer of risk. Under the IND framework, protocol amendments, information amendments, and safety reports each have specific regulatory meaning. A protocol amendment may change trial conduct. An information amendment may update nonclinical, CMC, clinical, or other information. A safety report may communicate an urgent risk signal. Regulatory affairs must know which pathway fits the content, because inaccurate submission classification can confuse FDA correspondence and weaken internal control. The eCFR table of contents for 21 CFR Part 312 identifies protocol amendments, information amendments, and IND safety reporting as distinct regulatory sections.
The best teams maintain a regulatory decision log. Every FDA question, meeting comment, safety signal, protocol change, CMC change, data issue, labeling decision, and commitment should have an owner, due date, rationale, affected documents, and closeout evidence. This protects the submission from organizational memory loss. It also helps future reviewers understand the history behind the package. That discipline belongs in clinical trial project management, clinical operations management, clinical trial sponsor oversight, and principal investigator responsibilities.
Communication style also matters. FDA correspondence should be direct, precise, respectful, and evidence-based. Internal communication should be brutally clear about risk. A regulatory affairs professional should never hide a weak area behind vague language. If safety exposure is limited, say exactly where. If the pivotal trial population is narrower than the proposed label, address it before reviewers do. If manufacturing comparability depends on specific bridging data, make that bridge visible. That is how scientific communication, medical science liaison discipline, medical monitor judgment, and regulatory affairs mastery come together.
5. Prevent the Regulatory Failure Modes That Delay Approval
Most IND/NDA problems are visible before they become submission problems. The warning signs are usually inconsistency, silence, and late discovery. Inconsistency appears when endpoint wording changes across the protocol, SAP, CSR, and labeling. Silence appears when teams avoid unresolved safety, CMC, or data integrity questions because nobody wants to slow momentum. Late discovery appears when regulatory operations finds missing documents after the content team believes the package is almost done.
One high-value habit is a submission risk register. The register should track major risks by module, owner, probability, impact, mitigation plan, decision deadline, and escalation path. For example, a safety risk may involve adverse events of special interest with uneven capture across studies. A data risk may involve unresolved query volume close to database lock. A CMC risk may involve stability data timing. A labeling risk may involve indication language that exceeds pivotal evidence. This kind of discipline draws from clinical trial resource allocation, risk-based monitoring, clinical trial budget management, and handling protocol deviations.
Another failure mode is poor source-to-submission traceability. Reviewers may ask why a claim was made, why a population was selected, why an analysis was used, or why a safety conclusion is reasonable. The team should be able to trace the answer from submission summary to study report to table to dataset to source process. That traceability depends on strong managing study documentation, CRA documentation techniques, clinical trial auditing, and site monitoring fundamentals.
Regulatory affairs professionals also need to challenge timeline fantasy. Teams often underestimate the time needed for final data cleaning, integrated summary drafting, cross-functional review, quality control, publishing, validation, submission, and response preparation. A rushed NDA can produce a long review cycle because the package forces reviewers to resolve questions the sponsor should have resolved internally. Better teams create milestone gates for draft completion, data finalization, medical review, statistical review, safety review, CMC review, labeling review, regulatory QC, technical validation, and executive approval.
Global strategy adds another layer. A sponsor pursuing multiple markets needs to watch for divergent regional expectations in clinical data, pediatric planning, risk management, labeling, safety reporting, and post-approval commitments. A single global dossier can support efficiency, but regional adaptations need control. A team using global regulatory compliance in pharmacovigilance, clinical research regulatory guidelines, clinical research ethics resources, and clinical trial market trends can avoid contradictory commitments that damage speed and credibility.
The final mastery point is mindset. IND/NDA submissions reward professionals who think like reviewers before the package reaches reviewers. They test claims. They hunt inconsistencies. They ask whether the protocol, data, safety profile, CMC package, and label tell the same story. They understand that a strong regulatory submission is not assembled at the end; it is earned through every decision made from preclinical planning through clinical conduct, database lock, integrated analysis, and lifecycle management.
6. FAQs About IND/NDA Submissions and Regulatory Affairs Mastery
-
An IND supports the legal and scientific path to study an investigational drug in humans, while an NDA supports the request to market a drug based on accumulated evidence. IND work focuses heavily on safe trial initiation, protocol quality, investigator readiness, CMC sufficiency for the development phase, and safety reporting. NDA work focuses on whether the total package proves safety, effectiveness, manufacturing quality, and labeling support. Professionals building careers in regulatory affairs, clinical regulatory specialist roles, IND strategy, and clinical trial sponsor responsibilities need to understand both.
-
NDA preparation should begin during development planning, not after pivotal trials finish. The team should define the target indication, endpoint hierarchy, statistical logic, safety monitoring approach, CMC evolution, labeling ambition, and data standards early enough to prevent avoidable gaps. If the NDA story starts after database lock, the team loses the chance to fix design-level weaknesses. The smartest preparation connects clinical trial protocol management, biostatistics, clinical data review, and pharmacovigilance safety monitoring.
-
Delays often come from inconsistent narratives, missing documents, unclear safety assessment, unresolved data issues, weak CMC readiness, poor amendment tracking, rushed labeling strategy, and eCTD publishing problems. Teams also lose time when responsibilities are unclear between sponsor, CRO, vendors, investigators, safety teams, data teams, and regulatory operations. Strong prevention depends on vendor management, clinical trial documentation, GCP compliance, and inspection readiness.
-
The highest-value skills are regulatory interpretation, evidence mapping, cross-functional leadership, clinical development literacy, safety judgment, CMC awareness, document control, eCTD awareness, and reviewer-style thinking. Regulatory affairs professionals should be able to translate scientific evidence into submission strategy while keeping teams honest about weaknesses. Career growth is stronger when professionals study regulatory affairs associate fundamentals, quality assurance careers, clinical compliance officer skills, and clinical research administrator leadership.
-
eCTD affects how reviewers navigate the submission, understand lifecycle updates, find evidence, and follow document relationships. A clean eCTD sequence reduces friction. A messy sequence can create confusion even when the underlying science is strong. FDA identifies eCTD as the standard format for many regulatory submissions to CDER and CBER, so regulatory operations capability is a core submission skill rather than a back-office detail. Teams should pair eCTD discipline with regulatory submission planning, clinical trial templates, and clinical research technology skills.
-
The fastest path is to study submissions as systems. Learn how protocol design affects data interpretation, how data quality affects labeling, how safety reporting affects risk-benefit assessment, how CMC changes affect review confidence, and how regulatory correspondence affects future commitments. Build practical fluency through clinical research certification, regulatory affairs career roadmaps, clinical research continuing education, and GCP-certified professional development. Regulatory mastery grows when every document is treated as part of one defensible evidence story.