
The Ultimate Guide to Getting Your Good Clinical Practice (ICH-GCP) Certification in South Dakota: Everything You Need to Know in 2026-27
South Dakota offers a distinctive route into clinical research through major health systems, community-based studies, rural trial delivery, pediatric research, oncology programs, and technology-supported participation. Candidates who pair South Dakota clinical research certification with ICH-GCP principles, clinical trial documentation skills, and site monitoring knowledge can compete for coordinator, regulatory, data, safety, and monitoring roles. Sanford Health and Avera both maintain active clinical research operations serving patients in South Dakota and the wider Midwest.
1. Why ICH-GCP Certification Matters in South Dakota in 2026-27
South Dakota’s clinical research market rewards professionals who can help trials operate across large geographic areas without weakening participant protection, documentation quality, or protocol control. Avera Research Institute conducts community-based research and clinical trials involving historically underserved populations, while its work includes pediatric research infrastructure in areas that have traditionally received lower levels of NIH funding. Sanford Health maintains a Sioux Falls clinical research center within a larger regional research network. These environments create opportunities for candidates trained in patient retention strategies, remote monitoring workflows, clinical trial safety reporting, and quality-focused study management.
The most expensive South Dakota trial problems rarely begin with obscure scientific questions. They begin with ordinary operational failures: an outdated consent form is used, a participant misses a safety assessment, a hospitalization is reported late, a source note fails to support an EDC entry, or a training record cannot prove that someone was qualified to perform a delegated task. Strong Good Clinical Practice education prepares you to identify those risks through protocol deviation analysis, SAE reporting procedures, investigator oversight principles, and clinical trial data integrity.
ICH E6(R3) raises the value of modern GCP training because the final FDA guidance emphasizes flexible, proportionate, risk-based approaches and accommodates innovation in trial design, conduct, and technology. Certification programs built around older terminology alone can leave candidates unprepared for quality-by-design thinking, computerized systems, decentralized trial elements, central monitoring, and critical-to-quality factors. South Dakota applicants should study risk-based monitoring strategies, clinical trial technology innovations, remote and on-site monitoring, and GCP compliance assessment alongside the core ethical principles.
Rural trial operations add another layer of professional responsibility. Long travel distances, weather disruptions, limited specialist availability, participant transportation barriers, outside laboratory records, local healthcare-provider involvement, and remote data collection can create more handoffs. Every handoff increases the need for clear ownership, contemporaneous documentation, reliable escalation, and careful reconciliation. Candidates who understand clinical trial timelines, patient education resources, site startup activities, and sponsor responsibilities can help prevent logistical complexity from becoming a compliance failure.
A certificate becomes valuable when it changes how you think. You should begin seeing each study activity as a controlled chain: the approved protocol defines what must occur; qualified personnel perform authorized duties; source records document what happened; EDC entries reflect those records; safety information reaches the correct parties; deviations receive assessment and follow-up; monitoring verifies critical processes; and essential documents preserve the evidence. This systems-level perspective connects clinical trial amendments, clinical data review, investigator site oversight, and project closeout procedures.
| Competency | What You Must Be Able to Do | South Dakota Pain Point Addressed | CCRPS Resource |
|---|---|---|---|
| GCP foundation | Explain participant protection, protocol compliance, data reliability, and documented oversight. | Prevents certification from becoming an unsupported resume claim. | GCP ethics and patient safety |
| State-specific positioning | Connect your training to rural, community, hospital, pediatric, and oncology research operations. | Prevents generic applications that overlook South Dakota’s research environment. | South Dakota clinical research certification |
| Informed consent | Verify approved version, signatures, dates, timing, comprehension, and re-consent requirements. | Reduces participant-rights violations and inspection exposure. | ethical conduct under GCP |
| Eligibility review | Trace every inclusion and exclusion decision to reliable source evidence. | Prevents ineligible enrollment and unsupported screening decisions. | trial data integrity |
| Visit-window control | Calculate protocol windows, anticipate travel barriers, and escalate scheduling risk early. | Reduces missed assessments across geographically dispersed populations. | trial timeline management |
| Source documentation | Create attributable, contemporaneous, complete, consistent, and retrievable records. | Prevents gaps between clinical care, source notes, and EDC data. | clinical data review |
| EDC accuracy | Enter source-supported information and resolve queries without inventing missing details. | Protects database quality and endpoint credibility. | clinical data integrity |
| Adverse events | Capture onset, severity, action taken, outcome, causality information, and follow-up. | Prevents incomplete safety records and delayed medical review. | adverse event compliance |
| Serious adverse events | Recognize seriousness criteria and trigger immediate escalation through required pathways. | Protects participants when urgent information surfaces remotely or after hours. | SAE reporting procedures |
| Protocol deviations | Document facts, assess impact, identify root cause, report appropriately, and implement prevention. | Stops repeat errors from being dismissed as isolated mistakes. | protocol deviation examples |
| Delegation | Confirm that each task matches documented authorization, qualifications, and training. | Prevents cross-trained rural teams from working outside assigned responsibilities. | investigator responsibilities |
| PI oversight | Keep the investigator informed about enrollment, safety, deviations, staff performance, and unresolved risks. | Prevents delegation from weakening investigator accountability. | site operations oversight |
| Regulatory files | Maintain current approvals, CVs, licenses, training records, logs, correspondence, and amendments. | Creates retrievable evidence for monitors, auditors, and inspectors. | clinical trial templates |
| Protocol amendments | Control approval, training, consent updates, implementation dates, and superseded materials. | Prevents different locations from following different protocol versions. | clinical trial amendments |
| Study startup | Track contracts, budgets, IRB approval, supplies, system access, training, and activation readiness. | Prevents premature enrollment before operational controls are ready. | startup checklist generator |
| Participant retention | Address travel, weather, visit burden, reminders, communication, and reimbursement delays. | Protects enrollment value after participants enter the study. | patient retention strategies |
| Recruitment quality | Screen accurately, explain study expectations clearly, and avoid pressure-based enrollment. | Reduces screen failures, early withdrawals, and trust damage. | patient education resources |
| Remote trial activities | Define who performs each task, where data originate, and how abnormal findings are escalated. | Prevents decentralized workflows from creating responsibility gaps. | virtual clinical trials |
| Monitoring readiness | Prepare consent, source, regulatory, safety, IP, and action-item records before review. | Reduces avoidable findings and wasted monitoring time. | GCP monitoring techniques |
| Risk-based monitoring | Prioritize participant safety, eligibility, consent, endpoints, and high-risk processes. | Prevents equal attention being given to issues with unequal consequences. | risk-based monitoring |
| Investigational product | Control receipt, storage, temperature, dispensing, returns, reconciliation, and destruction. | Protects accountability when products move through regional supply chains. | IND application guide |
| Technology literacy | Understand EDC, CTMS, eConsent, ePRO, eTMF, wearables, portals, and audit trails. | Prepares candidates for hybrid and technology-enabled studies. | clinical trial technology |
| Computerized-system control | Protect access, preserve audit trails, correct records properly, and avoid shared credentials. | Prevents convenience-driven practices from undermining data credibility. | data integrity responsibilities |
| Quality management | Identify critical risks early and build preventive controls into routine workflows. | Reduces dependence on late corrective action. | quality management strategies |
| Sponsor-site communication | Escalate issues with facts, impact, ownership, deadline, and proposed next action. | Prevents vague emails and unresolved action items. | sponsor responsibilities |
| Budget awareness | Track completed visits, screen failures, pass-through expenses, invoice triggers, and unpaid items. | Protects research programs operating with limited staffing and resources. | CRC budget management |
| Audit readiness | Ensure every critical activity can be reconstructed through reliable records. | Prevents teams from relying on memory when evidence is requested. | audit and inspection techniques |
| Closeout | Resolve queries, reconcile product, file final documents, close actions, and preserve records. | Stops long-running documentation gaps from surfacing at study end. | clinical trial closeout |
| Career targeting | Choose one primary pathway: coordination, monitoring, regulatory, data, safety, or project support. | Prevents scattered applications and unfocused credential collection. | certificate programs compared |
| Interview proof | Build a portfolio containing a consent checklist, visit tracker, deviation record, and escalation workflow. | Gives employers evidence beyond course completion. | GCP self-assessment |
2. What a Strong ICH-GCP Certification Program Must Teach
A worthwhile program should begin with the ethical and scientific purpose of GCP. Participants accept inconvenience, uncertainty, privacy risk, procedures, and potential side effects so researchers can answer important questions. Every site process must therefore protect informed choice, physical safety, confidentiality, and reliable evidence. Candidates should be able to connect ethical conduct in research with investigator responsibilities, adverse event compliance, and clinical research ethics resources.
Informed consent training must cover more than obtaining signatures. You should learn how to confirm that the correct IRB-approved version is being used, give the participant adequate time, answer questions within your role, document the discussion, preserve voluntariness, provide a copy, and recognize when new information requires re-consent. In a state where some participants may travel considerable distances for specialty care, teams must avoid allowing scheduling pressure to shorten the consent process. Review patient education resources, GCP patient-safety principles, site monitoring expectations, and protocol compliance guidance.
The course should develop operational judgment around investigator oversight and delegation. The principal investigator retains responsibility for trial conduct while appropriately qualified staff may perform assigned duties. A delegation log gains value through accurate dates, defined tasks, documented qualifications, and timely training. A signature alone cannot correct a situation where the delegated person lacked preparation or authority. Study PI site oversight, investigator-initiated trials, leadership and team management, and quality management systems to understand how oversight functions in practice.
Data training should explain the relationship between source records, case report forms, EDC entries, corrections, queries, external records, devices, laboratory systems, and audit trails. A coordinator must never fill a documentation gap through assumption. A CRA must distinguish a simple transcription error from a systemic process weakness. A data professional must understand the clinical meaning behind fields that appear administrative. Build these skills through clinical trial data verification, data integrity responsibilities, trial technology rankings, and clinical trial template resources.
Safety modules should train you to collect complete information and escalate promptly. A participant saying, “I went to the hospital last weekend,” creates an immediate need to determine dates, reason for admission, medical outcome, current status, treatment, available records, and investigator awareness. Staff should recognize seriousness criteria while leaving medical causality judgments to qualified personnel. Candidates targeting South Dakota research roles should master SAE definitions and timelines, pharmacovigilance practices, global safety compliance, and PV inspection preparation.
A modern certification must also address risk proportionality. Teams should devote greater control to consent, eligibility, safety, investigational product, primary endpoints, and other critical-to-quality processes. This approach requires thoughtful prioritization rather than mechanical completion of every task with equal intensity. FDA’s final E6(R3) guidance expressly incorporates flexible, risk-based approaches and innovation in technology and trial conduct. Candidates should therefore understand risk-based monitoring, remote monitoring techniques, trial technology systems, and quality-by-design thinking.
3. How to Choose Your South Dakota Certification and Build Job-Ready Proof
Begin by choosing the role you want your certification to support. Clinical Research Coordinator candidates need strength in consent, eligibility, visit windows, source documentation, EDC, patient communication, safety follow-up, and monitoring preparation. Clinical Research Associate candidates need protocol interpretation, consent verification, source review, issue escalation, site communication, and risk-based oversight. Regulatory candidates need version control, IRB submissions, essential documents, training records, and amendment implementation. Compare these paths through South Dakota clinical research certification, CRA monitoring mastery, regulatory affairs training, and certificate program comparisons.
Examine the course syllabus before enrolling. Look for ICH E6(R3), informed consent, investigator and sponsor responsibilities, protocol compliance, safety reporting, data governance, computerized systems, essential documents, monitoring, audits, quality management, and decentralized elements. A short multiple-choice course may satisfy a narrow workplace refresher while providing limited career preparation. A career-focused learner needs scenario practice and explanations that connect each rule to site consequences. Supplement the syllabus with FDA-focused GCP resources, IND application guidance, clinical trial amendments, and trial sponsor best practices.
Check how the program measures understanding. Useful assessments should force you to apply GCP to situations involving an outdated consent version, missing investigator review, a late SAE, an undocumented correction, an untrained staff member, or a missed protocol procedure. Scenario-based learning exposes weaknesses that definition-based quizzes can conceal. Use the GCP compliance self-assessment, protocol deviation examples, SAE reporting guide, and clinical trial data review guide to test yourself beyond the course exam.
Your training should produce a small professional portfolio. Create an informed-consent quality checklist, participant visit-window tracker, mock source note, protocol deviation record, corrective-action example, adverse-event escalation flow, training matrix, and monitoring action-item log. Remove all real patient information and label every sample as educational. These materials demonstrate workflow thinking during an interview and help you discuss study startup activities, site monitoring visits, clinical research templates, and project timeline control with concrete examples.
Translate your previous experience into trial risk control. Nursing and clinical care can demonstrate patient communication, medication knowledge, safety recognition, and documentation. Laboratory work can demonstrate specimen integrity, chain-of-custody discipline, equipment procedures, and quality control. Administrative work can support regulatory tracking, scheduling, confidential records, and deadline management. Pharmacy experience can support investigational product accountability and adverse-event awareness. Data experience can support EDC accuracy and reconciliation. Strengthen that translation with collaboration skills for research assistants, clinical trial budget management, trial safety monitoring, and data verification skills.
Avoid collecting certificates without a job-targeting strategy. Employers gain limited insight from a resume containing many course names and few examples of applied judgment. Choose one primary lane for the next 60 to 90 days, build role-specific proof, learn the vocabulary used in postings, and prepare scenarios aligned with that lane. Use the clinical research career map, clinical research salary tool, professional association directory, and online clinical research communities to focus your next move.
What Is Keeping You From a South Dakota Clinical Research Role?
Select the obstacle that is costing you the most progress. Your result will provide a focused corrective action.
4. Career Paths After ICH-GCP Certification in South Dakota
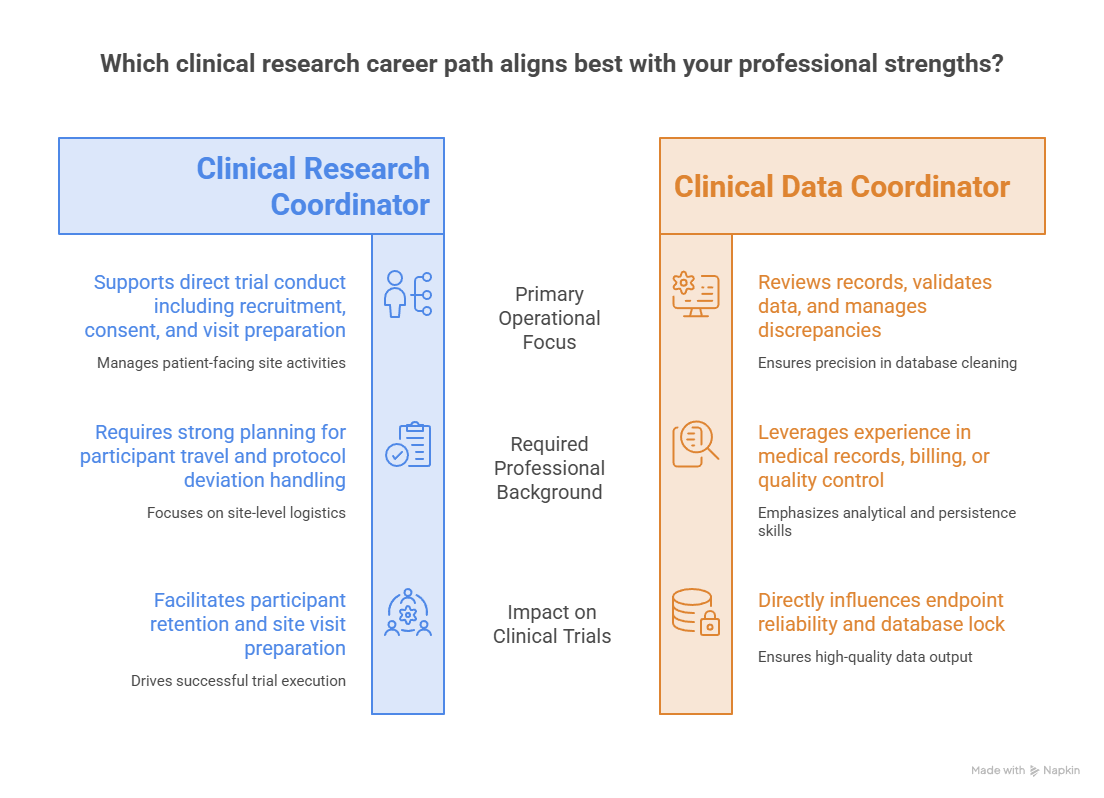
Clinical Research Coordinator and research-assistant roles offer the most direct exposure to trial conduct. These professionals may support recruitment, screening, consent, visit preparation, source documentation, laboratory coordination, EDC entry, safety follow-up, regulatory filing, and monitoring visits. In South Dakota, coordinators may also need strong planning for participants traveling from smaller communities. Study CRC patient retention, site visit preparation, CRC budget management, and protocol deviation handling to understand the full role.
Sanford Health provides a significant research environment in and around Sioux Falls. Its official clinical-trials materials list a Sioux Falls clinical research center and describe a system with more than 150 open clinical trials and 350 ongoing clinical studies. Sanford’s research portfolio includes adult and pediatric studies, oncology, genetics, rare and undiagnosed conditions, and other therapeutic areas. These operations can support careers involving clinical trial startup, clinical data verification, investigational product oversight, and monitoring visit management.
Avera provides another important South Dakota research setting. Its programs include cancer research, community-based research, genetics, maternal and child health, and participation in the NIH-supported IDeA States Pediatric Clinical Trials Network. Avera Research Institute maintains operations associated with Sioux Falls, Pierre, Rapid City, and other communities, which highlights the importance of distributed research coordination and culturally responsive participant engagement. Candidates can prepare through patient education resources, research ethics directories, participant retention strategy, and clinical trial technology.
Regulatory coordinator and clinical trial assistant roles suit candidates with strong document control, deadline tracking, and communication. The work can include IRB submissions, essential-document maintenance, investigator files, training records, delegation logs, safety correspondence, amendment implementation, system access, and site activation. A remote or hybrid regulatory role may also expand options for candidates living beyond Sioux Falls. Build readiness through clinical trial amendments, regulatory affairs mastery, trial startup checklists, and essential document templates.
Clinical data coordinator roles reward precision and persistence. Data professionals review records, enter or validate data, manage discrepancies, reconcile information, support database cleaning, and track queries. Their work can directly influence endpoint reliability, safety review, and database lock. Candidates with medical records, laboratory, analytics, insurance, billing, health-information, or quality-control experience may have transferable strengths. Develop them through clinical trial data review, data integrity responsibilities, technology innovation rankings, and trial timeline management.
Pharmacovigilance and safety roles may be accessible through regional employers or remote sponsor and CRO teams. These positions can involve adverse-event intake, case processing, narrative support, follow-up, medical coding, reconciliation, aggregate reporting support, and audit preparation. Nursing, pharmacy, biology, public health, medical terminology, and patient-support backgrounds can translate well when paired with strong GCP understanding. Strengthen your profile through clinical trial safety monitoring, global pharmacovigilance compliance, adverse event reporting requirements, and PV audit preparation.
The CRA pathway usually becomes more realistic after site, regulatory, data, or trial-assistant experience. Monitors must judge whether sites are protecting participants, following the protocol, producing credible data, controlling investigational products, resolving findings, and maintaining adequate oversight. South Dakota’s geographic realities also make remote-monitoring competence valuable. Candidates should master risk-based monitoring, on-site and remote visits, investigator meeting strategy, and CRA data verification before presenting themselves as monitoring-ready.
5. A 30-60-90 Day South Dakota ICH-GCP Certification Plan
During days 1 through 30, complete your ICH-GCP training and build a disciplined foundation. Study participant rights, informed consent, investigator oversight, sponsor responsibilities, protocol adherence, source documentation, safety reporting, deviations, essential documents, monitoring, computerized systems, and quality management. After each topic, write a scenario showing what failure looks like and how you would respond. Support this phase with GCP ethics and safety, investigator responsibility training, sponsor role guidance, and GCP compliance self-assessment.
Create a one-page command sheet by the end of week two. Include consent prerequisites, delegation requirements, ALCOA-related documentation principles, AE and SAE distinctions, deviation response, amendment control, monitoring purpose, and escalation structure. The sheet should help you answer interview questions without reciting memorized paragraphs. Add examples from protocol deviation guidance, serious adverse event procedures, clinical trial data integrity, and clinical amendment management.
During days 31 through 60, build your portfolio and rewrite your resume. Your portfolio should contain a consent review checklist, visit-window tracker, sample source note, deviation and CAPA example, safety escalation workflow, regulatory-document tracker, and monitoring action-item log. Every file should demonstrate clarity, ownership, deadlines, and escalation. Use the clinical trial template directory, trial startup checklist, monitoring visit guide, and quality management strategies as learning references.
Rewrite your employment history around transferable control skills. “Scheduled patients” can become evidence of time-sensitive appointment coordination and follow-up. “Maintained laboratory records” can demonstrate contemporaneous documentation and quality-control discipline. “Processed medical claims” can demonstrate discrepancy resolution and confidential data handling. “Managed medication inventory” can support accountability and reconciliation skills. Keep every statement truthful and specific. Use collaboration strategies for research assistants, CRC budget management, clinical data review skills, and patient retention practices to identify useful language.
During days 61 through 90, run a targeted application campaign. Create separate resume versions for Clinical Research Assistant, Assistant CRC, CRC, Regulatory Coordinator, Clinical Trial Assistant, Data Coordinator, and Safety Associate roles. Search Sioux Falls opportunities while also examining Pierre, Rapid City, regional health systems, universities, CROs, sponsors, and remote research operations. Use the clinical research career opportunity map, salary comparison tool, professional association directory, and online researcher communities to organize the search.
For every posting, identify the operational risk hidden behind each requirement. “Participant scheduling” signals visit windows and retention. “EDC experience” signals source-backed accuracy and query closure. “Regulatory maintenance” signals version control and inspection readiness. “Monitoring support” signals organized evidence and action-item follow-up. “Safety reporting” signals urgent escalation and complete information. Tailor your resume using clinical trial timeline management, GCP monitoring techniques, adverse event compliance, and regulatory affairs principles.
Prepare ten scenario answers before your first interview. Cover wrong consent version, missed protocol visit, late hospitalization report, EDC-source conflict, unavailable outside records, delegation mismatch, investigational product temperature excursion, delayed investigator review, amendment implementation, and repeat deviation. Structure each answer around immediate safety, fact collection, escalation, documentation, impact assessment, required reporting, and prevention. Refine your judgment through SAE reporting procedures, protocol deviation corrective actions, investigator oversight, and clinical research quality management.
6. FAQs About ICH-GCP Certification in South Dakota
-
Many employers expect evidence of GCP training because research personnel handle participant rights, protected health information, protocol procedures, safety information, source records, regulatory documents, and sponsor communication. Exact requirements vary by employer and role, and some organizations provide their own training after hiring. Completing certification beforehand strengthens your readiness for South Dakota clinical research roles, site monitoring support, GCP compliance responsibilities, and clinical data integrity work.
-
Course length depends on the provider, depth, assessment structure, and whether the program includes role-specific material. Completion speed should remain secondary to your ability to apply the principles. A candidate who finishes quickly and cannot manage an outdated consent form or late SAE remains underprepared. Pair your course with GCP ethics training, protocol deviation scenarios, SAE reporting practice, and trial documentation templates.
-
Relevant targets include Clinical Research Assistant, Assistant CRC, Clinical Research Coordinator, Regulatory Coordinator, Clinical Trial Assistant, Data Coordinator, Patient Recruitment Specialist, and Safety Associate. Experienced site professionals may later move toward CRA or project-management work. Choose a lane using clinical research certificate comparisons, CRC site visit guidance, CRA monitoring training, and pharmacovigilance best practices.
-
Healthcare, nursing, pharmacy, laboratory, public health, psychology, administration, data, and patient-support backgrounds can provide relevant skills. Your application must explain how those skills control clinical trial risks. Documentation accuracy, confidential-data handling, patient communication, specimen management, deadline tracking, inventory reconciliation, and safety escalation all have research value. Strengthen your bridge with research assistant communication skills, trial startup activities, data review competencies, and patient retention strategy.
-
Remote and hybrid opportunities can exist in regulatory operations, clinical trial assistance, data management, pharmacovigilance, centralized monitoring, project support, and participant follow-up. These roles still require secure systems, reliable documentation, clear communication, and disciplined escalation. Prepare through remote monitoring mastery, trial technology innovations, global safety compliance, and clinical trial timeline management.
-
CRA work requires applied monitoring judgment in addition to GCP knowledge. Employers may seek site experience, monitoring exposure, therapeutic-area familiarity, travel readiness, and strong written communication. A coordinator, regulatory professional, clinical trial assistant, or data specialist can use that experience as a bridge. Study risk-based monitoring strategies, on-site and remote monitoring, investigator meeting techniques, and clinical data verification.