
Quality Management Strategies for Clinical Research Projects
Clinical research quality fails when teams treat quality as a late-stage audit activity instead of a study design discipline. A strong project protects participants, preserves data credibility, reduces rework, and keeps sponsors ready for monitoring, inspection, submission, and publication. The strongest quality systems connect GCP compliance, clinical trial protocol management, risk-based monitoring, clinical trial data review, and inspection readiness into one working model.
1. Build Quality Into the Protocol Before the Study Begins
Quality management begins at protocol design because the protocol creates the conditions for every downstream success or failure. ICH E6(R3) emphasizes quality culture, proactive quality-by-design thinking, critical-to-quality factors, stakeholder engagement, and proportionate risk-based approaches in clinical trials. That means quality should be built into eligibility criteria, endpoint definitions, visit schedules, safety assessments, source documentation expectations, data collection tools, monitoring plans, and escalation pathways before the first participant is enrolled.
The first quality question should be painfully practical: can real sites execute this protocol correctly under normal workload pressure? A protocol can look scientifically elegant and still create errors if visit windows are narrow, procedures are duplicated, endpoints are vague, lab timing is unrealistic, or eligibility criteria require interpretation that coordinators cannot apply consistently. Strong teams pressure-test the protocol with CRC responsibilities, CRA monitoring expectations, principal investigator responsibilities, and clinical trial sponsor oversight before approval.
Critical-to-quality factors should be defined in plain operational language. For an oncology trial, the critical points may include eligibility confirmation, tumor assessment timing, adverse event grading, drug accountability, imaging review, and endpoint adjudication. For a decentralized trial, the critical points may include remote consent, device training, data transfer, participant identity verification, and missed-contact escalation. Every project should connect these factors to case report form design, primary and secondary endpoints, informed consent procedures, and adverse event reporting.
Protocol feasibility also protects budgets and timelines. Quality failures create hidden cost through re-consenting, query resolution, protocol deviations, site retraining, database lock delays, safety reconciliation, audit findings, and regulatory responses. A smart quality plan connects clinical trial budget management, clinical trial resource allocation, vendor management, and effective stakeholder communication early enough to prevent quality from becoming an emergency expense.
2. Create a Risk-Based Quality Management System That Works in Real Studies
Risk-based quality management should make teams faster, sharper, and more focused. FDA’s risk-based monitoring guidance states that sponsor oversight should focus on the most important aspects of study conduct and reporting to enhance human subject protection and clinical trial data quality. That principle should guide the entire project, from monitoring frequency to data review priorities, audit planning, site support, vendor oversight, and escalation.
A useful risk system starts with a study-level risk assessment. The team should identify what could threaten participant safety, data credibility, regulatory compliance, endpoint interpretability, recruitment integrity, and submission readiness. Each risk should receive a likelihood score, impact score, owner, mitigation plan, trigger point, review frequency, and escalation route. This connects risk-based monitoring strategies, clinical trial safety monitoring, project manager strategy, and clinical operations leadership into one active control system.
The next step is turning risk into monitoring behavior. High-risk eligibility criteria, consent version control, primary endpoint fields, investigational product accountability, SAE documentation, and critical lab values deserve targeted attention. Lower-risk fields can be reviewed through data trends, automated checks, and sample-based strategies. This approach protects quality resources from being consumed by low-value review. It also gives CRAs stronger logic for remote and on-site monitoring, site monitoring visits, clinical trial data verification, and GCP compliance for CRAs.
Risk review should happen throughout the study because project risk changes. A low-risk site can become high-risk after coordinator turnover. A manageable query trend can become a database lock threat. A minor vendor delay can become an endpoint completeness issue. A protocol amendment can change consent, CRF, database, training, monitoring, and statistical implications at once. FDA’s monitoring Q&A guidance discusses monitoring plans, monitoring results, and implementation of risk-based approaches for investigational studies. Strong teams use those concepts to keep quality management alive during execution.
The best risk systems are simple enough for teams to use under pressure. Overbuilt dashboards can become decorative. A practical system shows the top risks, trend direction, owner, next action, deadline, and escalation decision. Clinical research teams should connect this dashboard to protocol amendments, protocol deviations, clinical trial templates, and interactive GCP self-assessment so the team can move from observation to correction quickly.
3. Control Data Quality, Deviations, and CAPA Before Inspection Pressure Starts
Data quality depends on prevention, detection, correction, and proof of control. Prevention happens through strong protocol design, site training, CRF logic, edit checks, and source expectations. Detection happens through monitoring, data review, medical review, safety reconciliation, and statistical outlier checks. Correction happens through queries, retraining, CAPA, amendment, or process repair. Proof of control appears when the team can show that the issue was identified, assessed, corrected, and prevented from recurring.
The first practical step is designing a data quality plan around critical data. Primary endpoint fields, key secondary endpoints, eligibility criteria, consent data, dosing records, safety assessments, visit timing, and discontinuation reasons deserve higher attention than routine administrative fields. Data managers, CRAs, medical monitors, and statisticians should agree on which variables can damage interpretability. That conversation belongs beside biostatistics in clinical trials, sample size planning, placebo-controlled trial design, and clinical data manager career skills.
Deviation management is where many projects lose trust. A deviation log filled with repeated “site retrained” responses signals weak root-cause thinking. Good CAPA asks why the error happened, whether it affected participant safety or data integrity, whether other sites could have the same issue, what immediate correction is required, what preventive action will change behavior, and how effectiveness will be measured. This is where handling protocol deviations, protocol deviation definitions, clinical quality auditor skills, and clinical compliance officer guidance become operationally valuable.
CAPA effectiveness should be measured through evidence. If late AE entry caused repeated reporting issues, effectiveness may be shown by reduced AE entry lag after workflow change. If consent version errors happened after amendments, effectiveness may be shown by version-control checks across all active sites. If eligibility mistakes came from unclear lab cutoffs, effectiveness may be shown by corrected screening worksheet use and absence of repeat eligibility deviations. Quality teams should connect CAPA to managing regulatory documents, research compliance and ethics, managing study documentation, and clinical trial auditing.
Safety quality needs its own rhythm. AE and SAE review should be reconciled across source, EDC, safety database, medical review notes, narratives, and regulatory reporting records. Weak reconciliation can create contradictions between clinical data and pharmacovigilance outputs. That damages confidence during audits, inspections, aggregate reporting, and submission review. A strong project links what pharmacovigilance is, drug safety timelines, aggregate safety reports, and medical monitor adverse event review into the quality plan.
What is your biggest clinical research quality blocker right now?
Choose one. Your answer points to the quality strategy that will prevent the most rework.
4. Align Sponsors, Sites, Vendors, and Monitors Around One Quality Standard
Clinical research quality breaks when every group defines success differently. Sponsors may focus on timelines, sites may focus on visit execution, CROs may focus on deliverables, monitors may focus on findings, data teams may focus on queries, and safety teams may focus on reporting timelines. A real quality system aligns these groups around participant protection, protocol adherence, data credibility, and inspection-ready evidence.
Sponsor oversight remains central even when major work is outsourced. Vendors can run monitoring, data management, labs, imaging, safety processing, eTMF systems, and technology platforms, yet the project still needs unified quality ownership. The sponsor should define vendor quality expectations through scopes of work, quality agreements, metrics, escalation rules, communication cadence, issue management, audit rights, and deliverable acceptance criteria. This is where vendor management in clinical trials, clinical trial sponsor roles, clinical project manager strategy, and clinical trial resource allocation become quality tools.
Sites need quality expectations that make sense in real clinic flow. A site training deck alone rarely changes behavior. Coordinators need visit checklists, eligibility worksheets, consent version controls, AE prompts, documentation examples, escalation contacts, and rapid answers to ambiguity. Investigators need clear oversight expectations and enough visibility into deviations, safety trends, and participant risk. These controls connect naturally to CRC responsibilities, site monitoring visit steps, PI patient safety oversight, and GCP training requirements.
CRAs should function as quality interpreters, not finding collectors. A strong CRA recognizes patterns: a consent correction may signal version-control weakness; a missing lab time may signal workflow design failure; repeated EDC delays may signal staffing capacity; a drug accountability mismatch may signal delegation or training gaps. Monitoring reports should therefore explain risk, root cause, required action, and follow-up evidence. That level of monitoring maturity is built through CRA career development, senior CRA advancement, CRA inspection readiness, and CRA documentation techniques.
Quality alignment also requires clean governance. Study teams should hold structured quality review meetings that cover critical risks, major deviations, overdue actions, safety reconciliation, vendor performance, query aging, TMF completeness, recruitment quality, retention risk, and upcoming milestones. The meeting should end with decisions, owners, and dates. A discussion without accountable action creates the illusion of oversight. Better governance links clinical trial timeline management, clinical operations manager skills, clinical research project manager career paths, and clinical trial manager roadmaps.
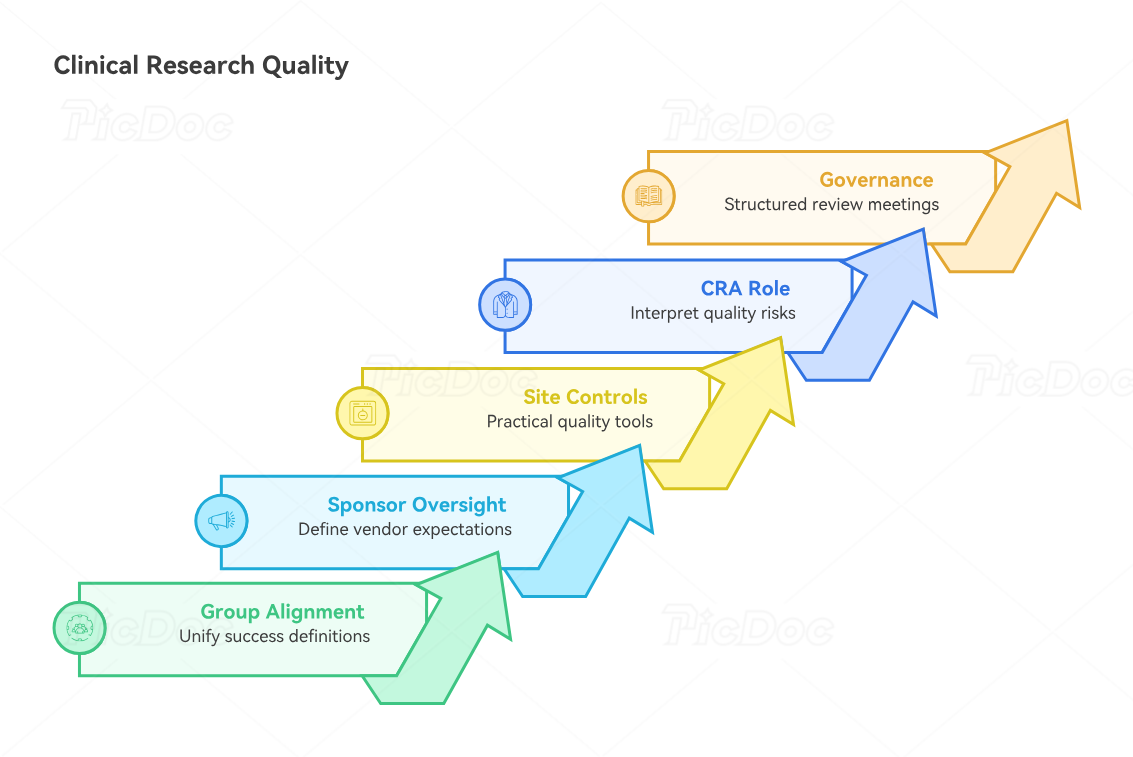
5. Use Metrics, Audits, and Quality Culture to Prevent Recurring Failures
Quality metrics should reveal behavior before failure becomes expensive. Useful clinical research metrics include consent error rate, major deviation frequency, deviation recurrence by root cause, query aging, critical query volume, visit window compliance, AE entry lag, SAE reporting timeliness, monitoring action item closure, TMF completeness by milestone, vendor deliverable timeliness, site activation delay reasons, participant discontinuation patterns, and database lock readiness.
The most valuable metrics are paired with thresholds and action rules. For example, a site with repeated visit-window deviations may trigger targeted retraining and workflow review. A vendor with repeated late deliverables may trigger escalation and corrective planning. A study with rising query aging may trigger data management triage. A high-risk endpoint with inconsistent source support may trigger targeted monitoring. Metrics should connect to clinical trial data verification, clinical data coordinator skills, lead clinical data analyst careers, and clinical research technology adoption.
Audits should test whether the quality system works under real conditions. An audit that only checks documents after a study is almost complete provides limited prevention value. Better audits target high-risk processes during active conduct: consent, eligibility, safety reporting, endpoint capture, vendor oversight, drug accountability, data flow, remote monitoring, and TMF controls. FDA’s clinical trial guidance page identifies agency guidance documents as current thinking on clinical trial conduct, good clinical practice, and human subject protection. Quality teams should use that kind of regulatory awareness to shape audit priorities.
Quality culture is built through how leaders respond to problems. If teams punish error reporting, problems go underground. If leaders reward speed while ignoring quality signals, staff learn to hide risk until deadlines force a crisis. If every CAPA closes with “retraining,” people stop believing in CAPA. Strong quality culture makes it safe to identify risk early and unacceptable to leave it unresolved. That mindset fits clinical research compliance, QA specialist career development, clinical quality auditor growth, and clinical compliance officer responsibilities.
The strongest projects prepare for inspection from day one. Inspection readiness means the story of the trial is clear, the evidence is complete, and the team can explain decisions. Essential documents should show who did what, when, why, under which version, with what oversight, and what happened when issues appeared. That story should be visible in the TMF, monitoring reports, safety files, deviation logs, CAPA records, training files, delegation logs, vendor records, and data review outputs. A project grounded in handling clinical trial audits, CRA inspection readiness, regulatory document management, and clinical trial documentation can defend itself without panic.
6. FAQs About Quality Management Strategies for Clinical Research Projects
-
Quality management in clinical research is the planned system used to protect participants, preserve data reliability, control protocol execution, manage risk, correct issues, and maintain inspection-ready evidence. It includes quality planning, risk assessment, monitoring, data review, deviation management, CAPA, vendor oversight, audit readiness, and continuous improvement. A strong system connects GCP compliance, clinical trial protocol management, clinical trial safety monitoring, and clinical trial auditing.
-
The most important risks usually involve participant safety, informed consent, eligibility, primary endpoint capture, AE and SAE reporting, protocol deviations, investigational product accountability, data integrity, vendor oversight, and essential document completeness. The exact risk profile depends on study phase, therapeutic area, design complexity, population vulnerability, endpoints, technology, and site experience. Teams should connect risk assessment to risk-based monitoring, serious adverse event reporting, endpoint clarity, and data monitoring committee roles.
-
Effective CAPA starts with root cause, not surface correction. The team should define what happened, why it happened, who or what was affected, whether the issue appears elsewhere, what immediate correction is needed, what preventive change will stop recurrence, and how effectiveness will be measured. Strong CAPA should reduce repeat errors. This requires disciplined use of protocol deviation guidance, handling protocol deviations, clinical quality auditor skills, and clinical compliance officer practices.
-
Risk-based monitoring improves quality by focusing oversight on the parts of the study that matter most for participant protection and data credibility. Instead of spreading equal effort across every field, teams focus on critical data, critical processes, risk signals, site performance, and emerging trends. This makes monitoring more meaningful and helps teams act before problems expand. The approach works best when paired with remote monitoring skills, site monitoring visit discipline, CRA GCP essentials, and clinical data review.
-
Clinical research teams should track metrics that reveal risk early: major deviations, deviation recurrence, consent errors, eligibility issues, AE entry lag, SAE reporting timeliness, query aging, critical query rates, visit-window compliance, missing endpoint data, monitoring action closure, vendor deliverable timeliness, TMF completeness, recruitment quality, retention patterns, and CAPA effectiveness. Metrics should trigger decisions, not passive reporting. They should connect to clinical trial data management, clinical research technology, project management strategy, and resource allocation.
-
A team becomes inspection-ready by keeping evidence current, traceable, and explainable throughout the study. That means clean delegation logs, training records, consent documentation, monitoring reports, deviation logs, CAPA files, safety records, vendor oversight evidence, TMF milestones, and data review decisions. The team should be able to explain the study story without hunting through disconnected files. Inspection readiness improves when teams use handling clinical trial audits, regulatory document control, clinical trial documentation methods, and CRA inspection readiness techniques.